Studios for interactive analysis
Studios allows users to host a variety of container images directly in Seqera Platform compute environments for analysis using popular environments including Jupyter (Python) and an R-IDE (R), Visual Studio Code IDEs, and Xpra remote desktops. Each Studio session provides a dedicated interactive environment that encapsulates the live environment. This guide explores how Studios integrates with your existing workflows, bridging the gap between pipeline execution and interactive analysis. It details how to set up and use each type of Studio, demonstrating a practical use case for each.
You will need the following to get started:
- At least the Maintain workspace user role to create and configure Studios.
- An AWS Batch compute environment (without Fargate) with sufficient resources (minimum: 2 CPUs, 8192 MB RAM).
- Valid credentials for your cloud storage account and compute environment.
- Data Explorer enabled in your workspace.
The scripts and instructions provided in this guide were tested on 24 February 2025. Library and package versions recommended here may become outdated and lead to unexpected results over time.
Jupyter: Python-based visualization of protein structure prediction data
Jupyter notebooks enable interactive analysis using Python libraries and tools. For example, Py3DMol is a tool used for visualizing and comparing structures produced by workflows such as nf-core/proteinfold, a bioinformatics best-practice analysis pipeline for protein 3D structure prediction. This section demonstrates how to create an AWS Batch compute environment, add the nf-core AWS megatests public proteinfold data to your workspace, create a Jupyter Studio, and run the provided Python script to produce interactive composite 3D images of the H1065 sequence.
This script and instructions can also be used to visualize the structures from nf-core/proteinfold runs performed with your own public or private data.
Create an AWS Batch compute environment
Studios require an AWS Batch compute environment. If you do not have an existing compute environment available, create one with the following attributes:
- Region: To minimize costs, your compute environment should be in the same region as your data. To browse the nf-core AWS megatests public data optimally, select eu-west-1.
- Provisioning model: Use On-demand EC2 instances.
- Studios does not support AWS Fargate. Do not enable Use Fargate for head job.
- At least 2 available CPUs and 8192 MB of RAM.
Add data using Data Explorer
For the purposes of this guide, add the proteinfold results (H1065 sequence) from the nf-core AWS megatests S3 bucket to your workspace using Data Explorer:
- From the Data Explorer tab, select Add cloud bucket.
- Specify the bucket details:
- Provider: AWS
- Bucket path:
s3://nf-core-awsmegatests/proteinfold/results-9bea0dc4ebb26358142afbcab3d7efd962d3a820 - A unique Name for the bucket, such as
nf-core-awsmegatests-proteinfold-h1065 - Credentials: Public
- An optional bucket Description
- Select Add.
To use your own pipeline data for interactive visualization, add the cloud bucket that contains the results of your nf-core/proteinfold pipeline run. See Add a cloud bucket for more information.
Create a Jupyter Studio session
From the Studios tab, select Add a Studio and complete the following:
- In the Compute & Data tab:
- Select your AWS Batch compute environment.
note
Studio sessions compete for computing resources when sharing compute environments. Ensure your shared compute environment has sufficient resources to run both your pipelines and Studio sessions.
- Optional: Enter CPU and memory allocations. The default values are 2 CPUs and 8192 MB memory (RAM).
- Mount data using Data Explorer: Mount the S3 bucket or directory path that contains the nf-core AWS megatests proteinfold data, or the work directory of your nf-core/proteinfold run.
- Select your AWS Batch compute environment.
- In the General config tab:
- Select the latest Jupyter container image template from the list.
- Optional: Enter a unique name and description for the Studio.
- Check Install Conda packages and paste the following into the YAML textfield:
channels:
- schrodinger
- conda-forge
- bioconda
dependencies:
- python=3.10
- conda-forge::libgl
- pip
- pip:
- biopython==1.85
- mdtraj==1.10.3
- py3dmol==2.4.2 - Select Add or choose to Add and start a Studio session immediately.
- If you chose to Add the Studio in the preceding step, select Connect in the options menu to open a Studio session in a new browser tab.
Visualize protein structures
The following Python script visualizes and compares protein structures produced by Alphafold 2 and ESMFold, creating a composite interactive 3D image of the two structures with contrasting colors. The script aligns mobile structures to reference structures, retrieves lists of C-alpha atoms from both structures, creates views for individual and combined structures, and creates an interactive view of the individual and combined structures using Py3DMol.
Run the following script in your Jupyter notebook to install the necessary packages and perform visualization:
Full Python script
import py3Dmol
from IPython.display import display
from Bio import PDB
from Bio.PDB import Superimposer
import numpy as np
# Keep file paths unchanged to visualize structures of the H1065 sequence in nf-core AWS megatests.
# Update file paths (to PDB files) to visualize structures of your own nf-core/proteinfold output data.
alphafold2_multimer_standard = "/workspace/data/nf-core-awsmegatests-proteinfold-h1065/mode_alphafold2_multimer/alphafold2/standard/H1065.alphafold.pdb"
esmfold_multimer = "/workspace/data/nf-core-awsmegatests-proteinfold-h1065/mode_esmfold_multimer/esmfold/H1065.pdb"
def align_structures(ref_pdb_path, mobile_pdb_path):
"""Align mobile structure to reference structure and return aligned coordinates"""
# Set up parser
parser = PDB.PDBParser()
# Load structures
ref_structure = parser.get_structure("reference", ref_pdb_path)
mobile_structure = parser.get_structure("mobile", mobile_pdb_path)
# Get lists of C-alpha atoms from both structures
ref_atoms = []
mobile_atoms = []
for model in ref_structure:
for chain in model:
for residue in chain:
if 'CA' in residue:
ref_atoms.append(residue['CA'])
for model in mobile_structure:
for chain in model:
for residue in chain:
if 'CA' in residue:
mobile_atoms.append(residue['CA'])
# Align structures using Superimposer
super_imposer = Superimposer()
super_imposer.set_atoms(ref_atoms, mobile_atoms)
super_imposer.apply(mobile_structure.get_atoms())
# Save aligned structure
io = PDB.PDBIO()
io.set_structure(mobile_structure)
aligned_pdb_path = "./"+mobile_pdb_path.split("/")[-1].replace('.pdb', '_aligned.pdb')
io.save(aligned_pdb_path)
return aligned_pdb_path
def create_structure_view(pdb_path, color, width=400, height=400, label=None):
"""Create a view for a single structure"""
view = py3Dmol.view(width=width, height=height)
with open(pdb_path, 'r') as f:
pdb_data = f.read()
view.addModel(pdb_data, "pdb")
view.setStyle({'model': -1}, {'cartoon': {'color': color}})
view.zoomTo()
if label:
view.addLabel(label, {
'position': {'x': 0, 'y': 0, 'z': 0},
'backgroundColor': color,
'fontColor': 'white'
})
return view
def visualize_structures(pdb1_path, pdb2_path):
# Align the second structure to the first
aligned_pdb2_path = align_structures(pdb1_path, pdb2_path)
# Create three separate views
view1 = create_structure_view(pdb1_path, 'blue', label="AlphaFold2")
view2 = create_structure_view(aligned_pdb2_path, 'darkgrey', label="ESMFold")
# Create combined view
view3 = py3Dmol.view(width=800, height=400)
# Load and display first structure (AlphaFold2)
with open(pdb1_path, 'r') as f:
pdb1_data = f.read()
view3.addModel(pdb1_data, "pdb")
view3.setStyle({'model': -1}, {'cartoon': {'color': 'blue'}})
# Load and display aligned second structure (ESMFold)
with open(aligned_pdb2_path, 'r') as f:
pdb2_data = f.read()
view3.addModel(pdb2_data, "pdb")
view3.setStyle({'model': 1}, {'cartoon': {'color': 'darkgrey'}})
# Set up the combined view
view3.zoomTo()
# Add labels for combined view
view3.addLabel("AlphaFold2", {'position': {'x': -20, 'y': 0, 'z': 0}, 'backgroundColor': 'blue', 'fontColor': 'white'})
view3.addLabel("ESMFold", {'position': {'x': 20, 'y': 0, 'z': 0}, 'backgroundColor': 'darkgrey', 'fontColor': 'white'})
return view1, view2, view3
# Visualize the structures
view1, view2, view3 = visualize_structures(alphafold2_multimer_standard, esmfold_multimer)
# Display all views
print("AlphaFold2 Structure:")
view1.show()
print("\nESMFold Structure:")
view2.show()
print("\nAligned Structures:")
view3.show()
Python script individual steps
-
Import libraries:
import py3Dmol
from IPython.display import display
from Bio import PDB
from Bio.PDB import Superimposer
import numpy as np -
Set up PDB file paths:
# Keep file paths unchanged to visualize structures of the H1065 sequence in nf-core AWS megatests.
# Update file paths (to PDB files) to visualize structures of your own nf-core/proteinfold output data.
alphafold2_multimer_standard = "/workspace/data/nf-core-awsmegatests-proteinfold-h1065/mode_alphafold2_multimer/alphafold2/standard/H1065.alphafold.pdb"
esmfold_multimer = "/workspace/data/nf-core-awsmegatests-proteinfold-h1065/mode_esmfold_multimer/esmfold/H1065.pdb" -
Load structures from the PDB files and retrieve lists of C-alpha atoms from both structures:
def align_structures(ref_pdb_path, mobile_pdb_path):
"""Align mobile structure to reference structure and return aligned coordinates"""
# Set up parser
parser = PDB.PDBParser()
# Load structures
ref_structure = parser.get_structure("reference", ref_pdb_path)
mobile_structure = parser.get_structure("mobile", mobile_pdb_path)
# Get lists of C-alpha atoms from both structures
ref_atoms = []
mobile_atoms = []
for model in ref_structure:
for chain in model:
for residue in chain:
if 'CA' in residue:
ref_atoms.append(residue['CA'])
for model in mobile_structure:
for chain in model:
for residue in chain:
if 'CA' in residue:
mobile_atoms.append(residue['CA']) -
Align structures using Superimposer:
# Align structures using Superimposer
super_imposer = Superimposer()
super_imposer.set_atoms(ref_atoms, mobile_atoms)
super_imposer.apply(mobile_structure.get_atoms())
# Save aligned structure
io = PDB.PDBIO()
io.set_structure(mobile_structure)
aligned_pdb_path = "./"+mobile_pdb_path.split("/")[-1].replace('.pdb', '_aligned.pdb')
io.save(aligned_pdb_path)
return aligned_pdb_path -
Create a view for a single structure:
def create_structure_view(pdb_path, color, width=400, height=400, label=None):
"""Create a view for a single structure"""
view = py3Dmol.view(width=width, height=height)
with open(pdb_path, 'r') as f:
pdb_data = f.read()
view.addModel(pdb_data, "pdb")
view.setStyle({'model': -1}, {'cartoon': {'color': color}})
view.zoomTo()
if label:
view.addLabel(label, {
'position': {'x': 0, 'y': 0, 'z': 0},
'backgroundColor': color,
'fontColor': 'white'
})
return view -
Create individual and combined structure views:
def visualize_structures(pdb1_path, pdb2_path):
# Align the second structure to the first
aligned_pdb2_path = align_structures(pdb1_path, pdb2_path)
# Create three separate views
view1 = create_structure_view(pdb1_path, 'blue', label="AlphaFold2")
view2 = create_structure_view(aligned_pdb2_path, 'darkgrey', label="ESMFold")
# Create combined view
view3 = py3Dmol.view(width=800, height=400)
# Load and display first structure (AlphaFold2)
with open(pdb1_path, 'r') as f:
pdb1_data = f.read()
view3.addModel(pdb1_data, "pdb")
view3.setStyle({'model': -1}, {'cartoon': {'color': 'blue'}})
# Load and display aligned second structure (ESMFold)
with open(aligned_pdb2_path, 'r') as f:
pdb2_data = f.read()
view3.addModel(pdb2_data, "pdb")
view3.setStyle({'model': 1}, {'cartoon': {'color': 'darkgrey'}})
# Set up the combined view
view3.zoomTo()
# Add labels for combined view
view3.addLabel("AlphaFold2", {'position': {'x': -20, 'y': 0, 'z': 0}, 'backgroundColor': 'blue', 'fontColor': 'white'})
view3.addLabel("ESMFold", {'position': {'x': 20, 'y': 0, 'z': 0}, 'backgroundColor': 'darkgrey', 'fontColor': 'white'})
return view1, view2, view3 -
Display interactive 3D structure views:
# Visualize the structures
view1, view2, view3 = visualize_structures(alphafold2_multimer_standard, esmfold_multimer)
# Display all views
print("AlphaFold2 Structure:")
view1.show()
print("\nESMFold Structure:")
view2.show()
print("\nAligned Structures:")
view3.show()
Interactive collaboration
To share a link to the running Studio session with collaborators inside your workspace, select the options menu for your Jupyter Studio session, then select Copy Studio URL. Using this link, other authenticated users can access the session directly to collaborate in real time.
R-IDE: Analyze RNASeq data and differential expression statistics
An R-IDE enables interactive analysis using R libraries and tools. For example, Shiny for R enables you to render functions in a reactive application and build a custom user interface to explore your data. The public data used in this section consists of RNA sequencing data that was processed by the nf-core/rnaseq pipeline to quantify gene expression, followed by nf-core/differentialabundance to derive differential expression statistics. This section demonstrates how to create a Studio to perform further analysis with these results from cloud storage. One of these outputs is web app that can be deployed for interactive analysis.
Create an AWS Batch compute environment
Studios require an AWS Batch compute environment. If you do not have an existing compute environment available, create one with the following attributes:
- Region: To minimize costs, your compute environment should be in the same region as your data. To browse the nf-core AWS megatests public data optimally, select eu-west-1.
- Provisioning model: Use On-demand EC2 instances.
- Studios does not support AWS Fargate. Do not enable Use Fargate for head job.
- At least 2 available CPUs and 8192 MB of RAM.
Add data using Data Explorer
For the purposes of this guide, add the nf-core AWS megatests S3 bucket to your workspace using Data Explorer:
- From the Data Explorer tab, select Add cloud bucket.
- Specify the bucket details:
- Provider: AWS
- Bucket path:
s3://nf-core-awsmegatests - A unique Name for the bucket, such as
nf-core-awsmegatests - Credentials: Public
- An optional bucket Description
- Select Add.
To use your own pipeline data for interactive analysis, add the cloud bucket that contains the results of your nf-core/differentialabundance pipeline run. See Add a cloud bucket for more information.
Create an R-IDE Studio session
From the Studios tab, select Add a Studio and complete the following:
- In the Compute & Data tab:
- Select your AWS Batch compute environment.
note
Studio sessions compete for computing resources when sharing compute environments. Ensure your compute environment has sufficient resources to run both your pipelines and Studio sessions.
- Optional: Enter CPU and memory allocations. The default values are 2 CPUs and 8192 MB memory (RAM).
- Mount data using Data Explorer: Mount the nf-core AWS megatests S3 bucket, or the directory path that contains the results of your nf-core/differentialabundance pipeline run.
- Select your AWS Batch compute environment.
- In the General config tab:
- Select the latest R-IDE container image template from the list.
- Optional: Enter a unique name and description for the Studio.
- Select Add or choose to Add and start a Studio session immediately.
- If you chose to Add the Studio in the preceding step, select Start in the options menu, then Connect to open a Studio session in a new browser tab when it is running.
Configure environment and explore data in a web app
The following R script installs and configures the prerequisite packages and libraries to deploy ShinyNGS, a web application created by members of the nf-core community to explore genomic data. The script also downloads the RDS file from nf-core AWS megatests to use as input data for the app's various plots, heatmaps, and tables. To use your own nf-core/rnaseq and nf-core/differentialabundance results, modify the script as instructed in step 2 below:
R script individual steps
-
Configure the R-IDE session with installed packages, including ShinyNGS:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(version = "3.20", ask = FALSE)
BiocManager::install(c("SummarizedExperiment", "GSEABase", "limma"))
install.packages(c("devtools", "matrixStats", "rmarkdown", "markdown"))
install.packages("shiny", repos = "https://cran.rstudio.com/")
devtools::install_version("cpp11", version = "0.2.1", repos = "http://cran.us.r-project.org")
devtools::install_github('pinin4fjords/shinyngs', upgrade_dependencies = FALSE) -
Download the RDS file from nf-core AWS megatests or your own nf-core/differentialabundance results (see Shiny app from the nf-core documentation for file details):
# For nf-core AWS megatests
download.file("https://nf-core-awsmegatests.s3-eu-west-1.amazonaws.com/differentialabundance/results-3dd360fed0dca1780db1bdf5dce85e5258fa2253/shinyngs_app/study/data.rds", 'data.rds')
# For your nf-core/differentialabundance results, replace the URL with your RDS file URL)
download.file("https://bucket.s3-region.amazonaws.com/differentialabundance/results/shinyngs_app/study-name/data.rds", 'data.rds') -
Import libraries, read your RDS data, and launch the app:
library(shinyngs)
library(markdown)
esel <- readRDS("data.rds")
app <- prepareApp("rnaseq", esel)
shiny::shinyApp(app$ui, app$server)
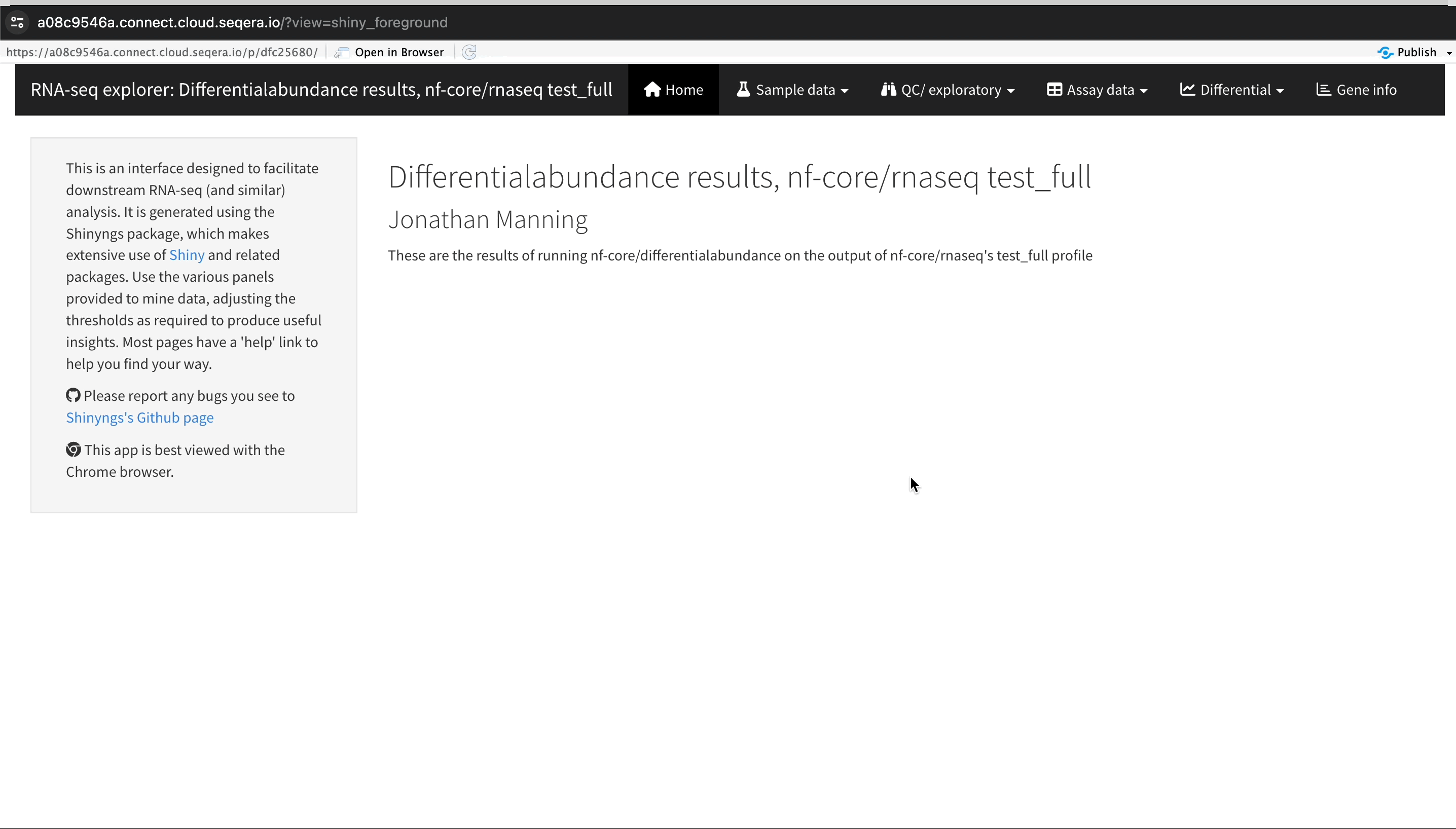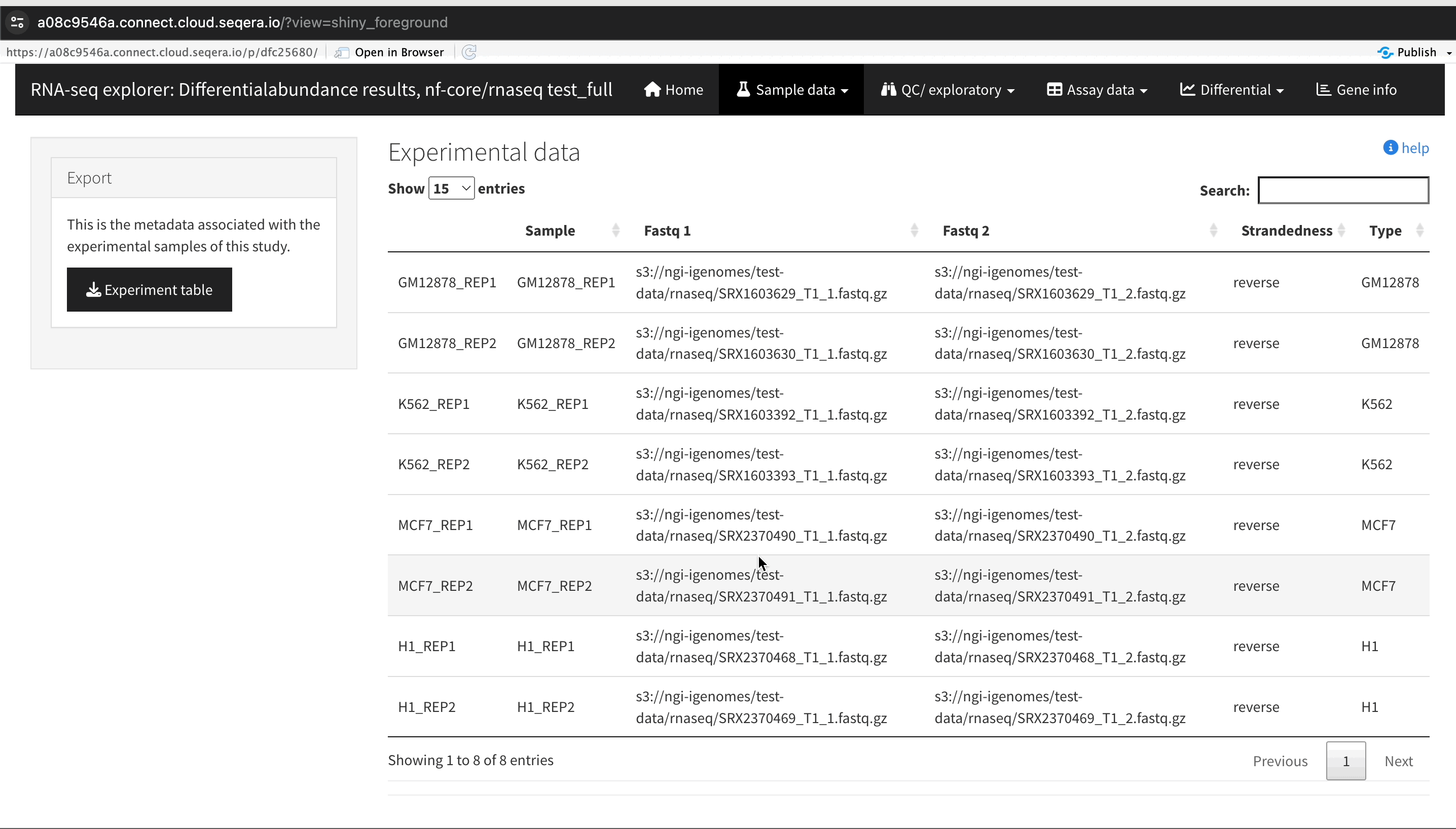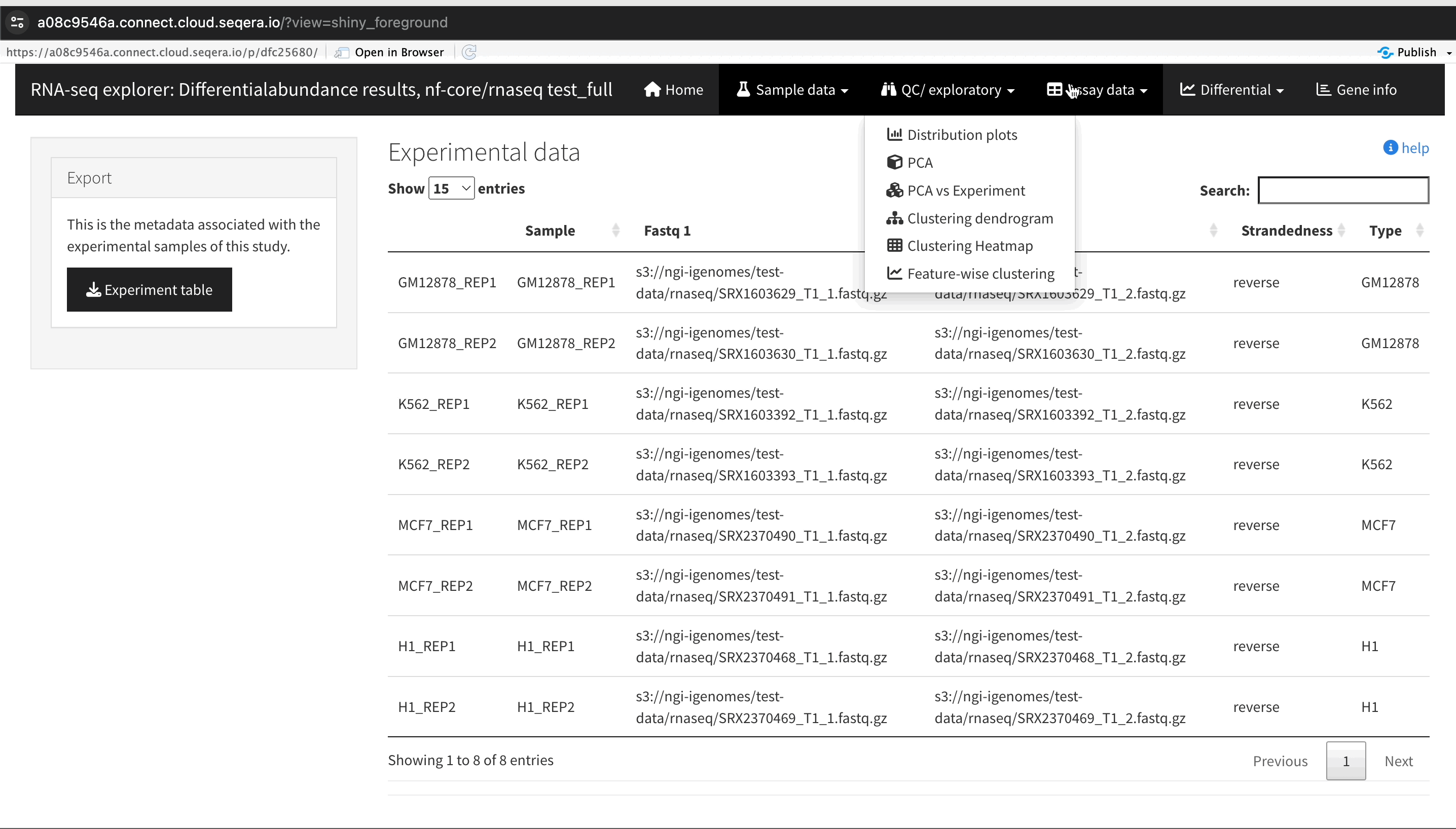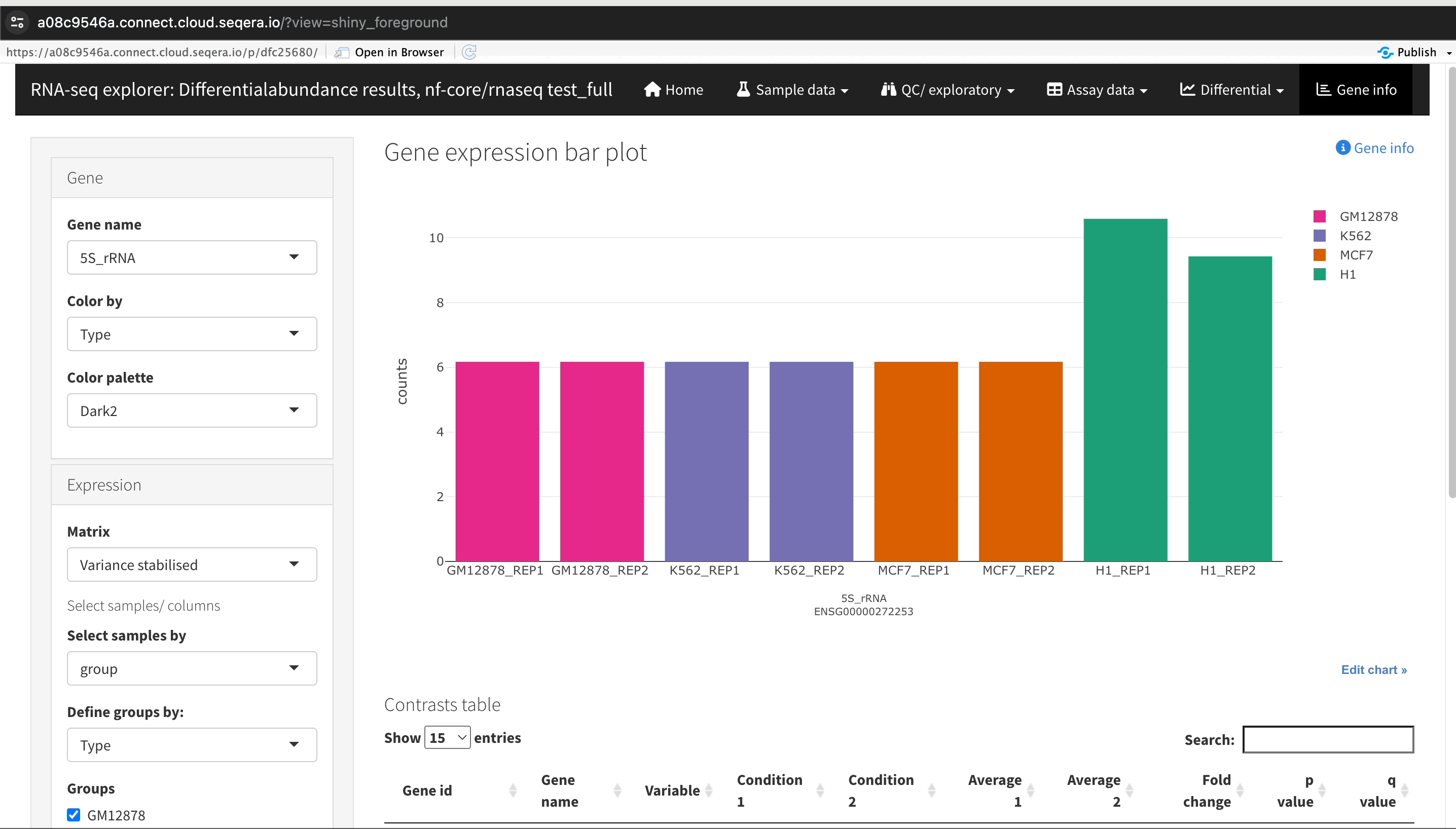
Interactive collaboration
To share a link to the running session with collaborators inside your workspace, select the options menu for your R-IDE session, then select Copy Studio URL. Using this link, other authenticated users can access the session directly to collaborate in real time.
Xpra: Visualize genetic variants with IGV
Xpra provides remote desktop functionality that enables many interactive analysis and troubleshooting workflows. One such workflow is to perform genetic variant visualization using IGV desktop, a powerful open-source tool for the visual exploration of genomic data. This section demonstrates how to add public data from the 1000 Genomes project to your workspace, set up an Xpra environment with IGV desktop pre-installed, and explore a variant of interest.
Create an AWS Batch compute environment
Studios require an AWS Batch compute environment. If you do not have an existing compute environment available, create one with the following attributes:
- Region: To minimize costs, your compute environment should be in the same region as your data. To browse the 1000 Genomes public data optimally, select us-east-1.
- Provisioning model: Use On-demand EC2 instances.
- Studios does not support AWS Fargate. Do not enable Use Fargate for head job.
- At least 2 available CPUs and 8192 MB of RAM.
Add data using Data Explorer
Add the 1000 Genomes S3 bucket to your workspace using Data Explorer:
- From the Data Explorer tab, select Add cloud bucket.
- Specify the bucket details:
- Provider: AWS
- Bucket path:
s3://1000genomes - A unique Name for the bucket, such as
1000G - Credentials: Public
- An optional bucket Description
- Select Add.
To use your own data for interactive analysis, see Add a cloud bucket for instructions to add your own public or private cloud bucket.
Create an Xpra Studio session
From the Studios tab, select Add a Studio and complete the following:
- In the Compute & Data tab:
- Select your AWS Batch compute environment.
note
Studio sessions compete for computing resources when sharing compute environments. Ensure your compute environment has sufficient resources to run both your pipelines and Studio sessions.
- Optional: Enter CPU and memory allocations.
- Mount the 1000 Genomes S3 bucket you added previously using Data Explorer.
- Select your AWS Batch compute environment.
- In the General config tab:
- Select the latest Xpra container image template from the list.
- Optional: Enter a unique name and description for the Studio.
- Check Install Conda packages and paste the following into the YAML textfield:
channels:
- conda-forge
- bioconda
dependencies:
- igv
- samtools
- Select Add or choose to Add and start a session immediately.
- If you chose to Add the Studio in the preceding step, select Connect in the options menu to open a session in a new browser tab.
View variants in IGV desktop
- In the Xpra terminal, run
igvto open IGV desktop. - In IGV, change the genome version to hg19.
- Select File, then Load from file, then navigate to
/workspace/data/xpra-1000Genomes/phase3/data/HG00096/high_coverage_alignmentand select the.baifile, as shown below: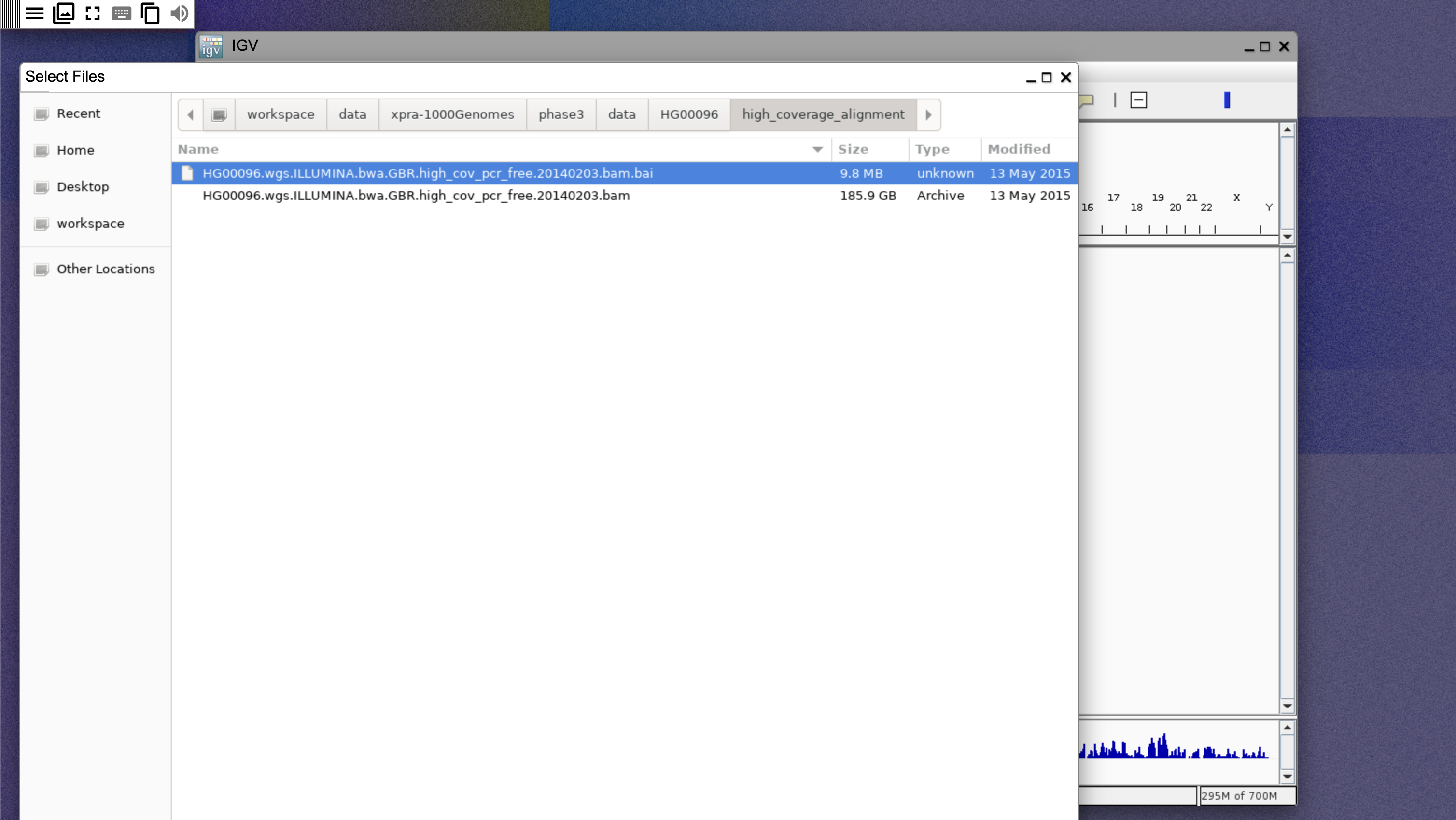
- Search for PCSK9 and zoom into one of the exons of the gene. A coverage graph and reads should be shown, as below:
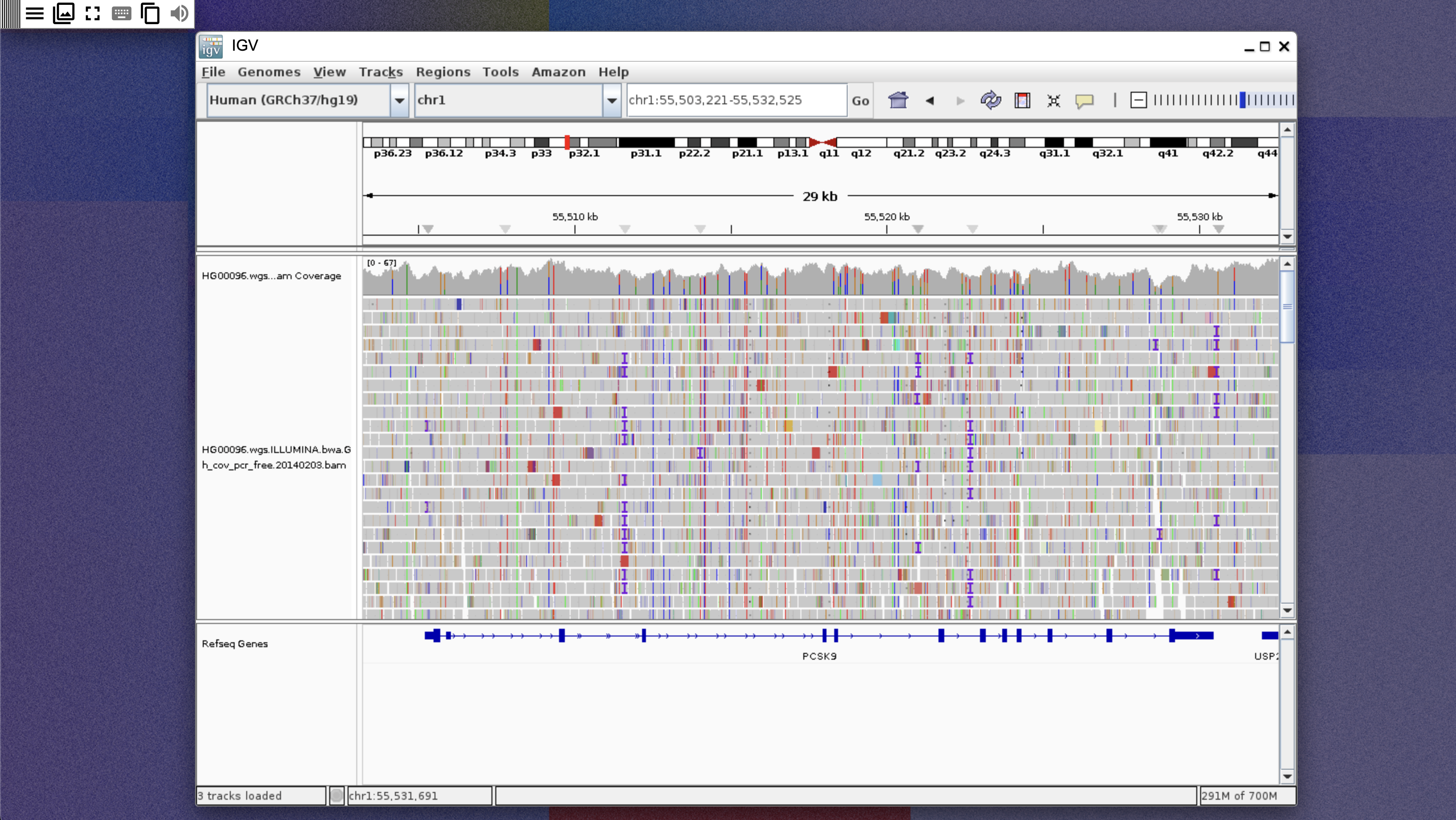
Interactive collaboration
To share a link to the running session with collaborators inside your workspace, select the options menu for your Xpra session, then select Copy Studio URL. Using this link, other authenticated users can access the session directly to collaborate in real time.
VS Code: Create an interactive Nextflow development environment
Using Studios and Visual Studio Code allows you to create a portable and interactive Nextflow development environment with all the tools you need to develop and run Nextflow pipelines. This section demonstrates how to set up a VS Code Studio with Conda and nf-core tools, add public data and run the nf-core/fetchngs pipeline with the test profile, and create a VS Code project to start coding your own Nextflow pipelines. The Studio includes the Nextflow VS Code extension, which makes use of the Nextflow language server to provide syntax highlighting, code navigation, code completion, and diagnostics for Nextflow scripts and configuration files.
Create an AWS Batch compute environment
Studios require an AWS Batch compute environment. If you do not have an existing compute environment available, create one with the following attributes:
- Region: To minimize costs, your compute environment should be in the same region as your data. To use the iGenomes public data bucket that contains the nf-core/fetchngs
testprofile data, select eu-west-1. - Provisioning model: Use On-demand EC2 instances.
- Studios does not support AWS Fargate. Do not enable Use Fargate for head job.
- At least 4 available CPUs and 16384 MB of RAM.
Add data using Data Explorer
The nf-core/fetchngs pipeline uses data from the NGI iGenomes public dataset for its test profile. To add this data to your workspace:
- From the Data Explorer tab, select Add cloud bucket.
- Specify the bucket details:
- Provider: AWS
- Bucket path:
s3://ngi-igenomes/test-data/ - A unique Name for the bucket, such as
ngi-igenomes-test-data - Credentials: Public
- An optional bucket Description
- Select Add.
Create a VS Code Studio session
From the Studios tab, select Add a Studio and complete the following:
- In the Compute & Data tab:
- Select your AWS Batch compute environment.
note
Studio sessions compete for computing resources when sharing compute environments. Shared compute environments must have sufficient resources to run both your pipelines and Studio sessions.
- Allocate at least 4 CPUs and 16384 MB RAM.
- Mount data using Data Explorer: To run nf-core/fetchngs with the
testprofile, mount the NGI iGenomes S3 bucket you added previously. Mount any other data directories you need to run and code your own Nextflow pipelines.
- Select your AWS Batch compute environment.
- In the General config tab:
- Select the latest VS Code container image template from the list.
- Optional: Enter a unique name and description for the Studio.
- Check Install Conda packages and paste the following into the YAML textfield:
channels:
- conda-forge
- bioconda
- anaconda
dependencies:
- nf-core
- conda
- Select Add or choose to Add and start a Studio session immediately.
- If you chose to Add the Studio in the preceding step, select Connect in the options menu to open a Studio session in a new browser tab.
- Once inside the Studio session, run
code .to use the clipboard.
See User and workspace settings if you wish to import existing VS Code configuration and preferences to your Studio session's VS Code environment.
Run nf-core/fetchngs with Conda
Run the following Nextflow command to run nf-core/fetchngs with Conda:
nextflow run nf-core/fetchngs -profile test,conda --outdir ./nf-core-fetchngs-conda-out -resume
Write a Nextflow pipeline with nf-core tools
- Run
nf-core pipelines createto create a new pipeline. Choose which parts of the nf-core template you want to use. - Run
code [your new pipeline]to open the new pipeline as a project in VSCode. This allows you to code your pipeline with the help of the Nextflow language server and nf-core tools.
Interactive collaboration
To share a link to the running session with collaborators inside your workspace, select the options menu for your VS Code Studio session, then select Copy Studio URL. Using this link, other authenticated users can access the session directly to collaborate in real time.