Writing New Modules
Introduction
Writing a new module can at first seem a daunting task. However, MultiQC has been written (and refactored) to provide a lot of functionality as common functions.
Provided that you are familiar with writing Python and you have a read through the guide below, you should be on your way in no time!
If you have any problems, feel free to contact the author - details here: @ewels
Philosophical concepts
These points are important and worth understanding early on. Get this stuff right, and your pull-request is much more likely to be merged quickly!
Don't add everything
MultiQC was designed to summarise tool outputs. An end-user should be able to visually scan the report and spot any outlier samples, then go to the underlying tool to look at those samples in more detail.
MultiQC is not designed to replicate every single metric from a tool. Doing so makes the report difficult to read and digest quickly for many samples. Module additions that add huge quantities of metrics to reports will be asked to slim down.
No images
MultiQC doesn't know how many samples it will need to handle for a report, and as such every module should work with anything from 1-1000 samples.
With images, you can't have more than a couple before the report is unusable. Worse, the file size will bloat the HTML file and it will crash the browser surprisingly fast. It's not accessible and the data cannot be exported into multiqc_data for downstream use.
Plots should be recreated within MultiQC by parsing the raw data and generating dynamic plots instead.
I almost never merge modules that include images into reports. If you really need images in your report, you can do this either via Custom Content or an unofficial plugin module. Feel free to discuss on the community forum if you think that your case is an exception. There have been one or two in the past.
One at a time
Please try to keep contributions as atomic as possible. In other words, one module = one pull request.
Don't be afraid to break things up into separate pull-requests coming from different branches. Just mention this in the PR comment so that it's clear which order they need to be merged in.
Avoid optimising too much
When you are writing a module that generates many similar plots, or table columns, or sections, it can be tempting to write nice efficient code that avoids duplicating these efforts. This is problematic for two reasons:
- It's bespoke to that module, so more difficult to maintain and comprehend
- Almost every table column, section and plot should have significant customisation. Descriptions, colour schemes, help text and more. Heavily optimised code will often need a lot of refactoring to pack this in.
It's usually better to copy and paste a bit in these cases. The code is then easier to understand and easier to customise.
Colour matters
The emphasis for MultiQC reports is to allow people to quickly scan and spot outlier samples. The core of this is data visualisation.
Especially when creating tables, make sure that you think about the colour scheme for every single column.
- Ensure that adjacent columns do not share the same colour scheme
- Makes long tables easier to follow
- Allows fast recognition of columns for regular users
- Think about what the colours suggest
- For example, if a large value is a bad thing (eg. percent duplication), use a strong red colour for large values
- If values are centred around a point (eg.
0), use a diverging colour scheme. Values close to the centre will have a weak colour and those at both ends of the distribution will be strongly coloured. - This allows rapid understanding without a lot of thought
This usually comes up for tables, but you can also think about it for bar plots.
Core modules / plugins
New modules can either be written as part of MultiQC or in a stand-alone plugin. If your module is for a publicly available tool, please add it to the main program and contribute your code back when complete via a pull request.
If your module is for something very niche, which no-one else can use, you can write it as part of a custom plugin. The process is almost identical, though it keeps the code bases separate. For more information about this, see the docs about MultiQC Plugins below.
Strict mode validation
MultiQC has been developed to be as forgiving as possible and will handle lots of invalid or ignored code. Even if a module raised an unexpected exception, MultiQC will log that error, and continue running.
This is useful most of the time, but can be difficult when writing new MultiQC
modules (especially during pull-request reviews). To help with this, you can run
MultiQC with the --strict flag. It will give explicit warnings about anything that
is not optimally configured, and will also make MultiQC exit early if a module crashed.
For example:
multiqc --strict test-data
Note that the automated MultiQC continuous integration testing runs in this mode, so you will need to pass all lint tests for those checks to pass. This is required for any pull-requests.
You can alternatively enable the strict mode using an environment variable:
export MULTIQC_STRICT=true
Or set it in the config:
# In multiqc_config.yaml
strict: True
Static code analysis
MultiQC uses type hints and static code analysis with mypy to prevent bugs.
Mypy is run on the entire codebase using a GitHub Actions job,
however, you can run it locally to check your changes before pushing them. In order to
do that, install MultiQC in the dev mode, which will bring mypy along with additional pluginsL
pip install -e .[dev]
Then run the following command to check your module:
mypy multiqc/modules/your_module
Fix any problems that mypy finds before submitting your pull request.
For a more convenient development experience, you can consider installing a mypy plugin for your editor. Both VS Code and PyCharm have plugins that can highlight type errors in your code as you write it.
Code formatting
MultiQC code base is also checked for consistency and formatting.
Everyone has their own preferences when it comes to writing any code, both in the methods
used but also with simple things like whitespace and whether to use " or '.
When reviewing code contributions in pull-requests, these variations in coding style
introduce an additional mental overhead. Inconsistent code style across the package
also makes it harder for newcomers to get into the code.
Code formatting / linting tools are able to assess files in many different languages and check that a set of "soft" formatting rules are adhered to, to enforce code consistency. Better still, many of these tools can automatically change the formatting so that developers can write code in whatever style they prefer and defer this task to automation.
Much like source control, gloves in a lab, and wearing a seatbelt, code formatters and code linting is an annoying inconvenience at first for most people which in time becomes an indispensable tool in the maintenance of high quality software.
MultiQC uses a range of tools to check the code base. The main two code formatters are:
The easiest way to work with these is to install editor plugins that run the tools every time you save a file. For example, Visual Studio Code has built-in support for Ruff and plugins for Prettier.
Other style considerations
-
We use modern Python 3, thus:
- Always use f-strings (e.g.
f"{var}") over the legacy"{var}".format()calls. - Use double quotes for strings.
- Built-in
dictpreserve order, thus most of the time you don't need to useOrderedDict. - Avoid unnecessary
__future__imports.
- Always use f-strings (e.g.
-
Unless a Python file is located in the root
scriptsdirectory, it must NOT have shebang lines like#!/usr/bin/env python.
Prek
MultiQC uses prek to test your code when you open a pull-request. Prek is a faster, Rust-based drop-in replacement for pre-commit.
It's recommended that you install it yourself in your MultiQC clone directory:
pip install prek # install the tool
prek install # set up prek in the MultiQC repository
This will then automatically run all code checks on the files you have edited when you create a commit. Prek cancels the commit if anything fails - sometimes it will have fixed files for you, in which case just add them and try to commit again. Sometimes you will need to read the logs and fix the problem manually.
Automated continuous integration tests will run using GitHub Actions to check that all files pass the above tests. If any files do not, that test will fail giving a red ❌ next to the pull request.
Make sure that your configuration is working properly and that you're not changing loads of files that you haven't worked with. Pull-requests will not be merged with such changes.
These tools should be relatively easy to install and run, and have integration with the majority of code editors. Once set up, they can run on save, and you'll never need to think about them again.
Initial setup
MultiQC file structure
The source code for MultiQC is separated into different folders. Most of the files you won't have to touch - the relevant files that you will need to edit or create follow the structure below:
├── docs
├── multiqc
│ ├── modules
│ | └── <your_module>
│ │ ├── __init__.py
│ │ ├── <your_module>.py
│ │ └── tests
│ │ ├── __init__.py
│ │ └── test_<your_module>.py
│ └── search_patterns.yaml
│ └── config_defaults.yaml
└── pyproject.toml
These files are described in more detail below.
Submodule
MultiQC modules are Python submodules - as such, they need their own
directory in multiqc/ with an __init__.py file. The directory should
share its name with the module. To follow common practice, the module
code itself usually then goes in a separate python file (also with the same
name, i.e. multiqc/bismark/bismark.py) which is then imported by the __init__.py file with:
from .mymodule import MultiqcModule
__all__ = ["MultiqcModule"]
Entry points
Once your submodule files are in place, you need to tell MultiQC that they
are available as an analysis module. This is done within pyproject.toml using
entry points.
In pyproject.toml you will see some code that looks like this:
[project.entry-points."multiqc.modules.v1"]
mymodule = "multiqc.modules.mymodule:MultiqcModule"
Copy one of the existing module lines and change it to use your module name. The order is irrelevant, so stick to alphabetical if in doubt. Once this is done, you will need to update your installation of MultiQC:
pip install -e .
MultiQC config
So that MultiQC knows what order modules should be run in, you need to add your module to the core config file.
In multiqc/config_defaults.yaml you should see a list variable called
module_order. This contains the name of modules in order of precedence. Add your
module here in an appropriate position.
Tests
Tests are written for pytest, and placed in the tests/
subdirectory within the module directory.
MultiQC has a blanket test that just checks that each module didn't crash when being run on the corresponding data in test-data, and added something into the report. However, users are encouraged to write more comprehensive tests that take the specific module logic into account. For some examples, consider checking:
- the samtools flagstat
test that verifies some logic in the
flagstatsubmodule of thesamtoolsmodule; - the picard tools test that checks that every submodule for each Picard tool worked correctly.
MultiqcModule Class
If you've copied one of the other entry point statements, it will have ended
in :MultiqcModule - this tells MultiQC to try to execute a class or function
called MultiqcModule.
To use the helper functions bundled with MultiQC, you should extend this
class from multiqc.modules.base_module.BaseMultiqcModule in your
module code file (i.e. multiqc/modname/modname.py). This will give
you access to a number of functions on the self namespace. For example:
from multiqc.base_module import BaseMultiqcModule
class MultiqcModule(BaseMultiqcModule):
def __init__(self):
super().__init__(
name="My Module",
anchor="mymodule",
href="https://www.awesome_bioinfo.com/mymodule",
info="Example analysis module used for writing documentation.",
doi=["01.2345/journal/abc123", "01.2345/journal/abc124"],
)
The __init__ variables are used to create the header, URL link,
analysis module credits and description in the report.
Markdown support in info
The info parameter supports rich markdown formatting. For example:
super().__init__(
name="My Advanced Module",
anchor="myadvancedmodule",
href="https://www.awesome_bioinfo.com/myadvancedmodule",
info="This module performs **quality assessment** of sequencing data.\n\n"
"Features:\n"
"- Quality score distribution\n"
"- Read length analysis\n"
"- *Fast* processing\n\n"
"See the [documentation](https://www.awesome_bioinfo.com/docs) for more details.",
doi="01.2345/journal/abc123",
)
The available arguments when initialising a module as follows:
- `name` - Name of your module
- `anchor` - A HTML-safe anchor that will be used after the `#` in the URL
- `href` - Link(s) to the homepage for the tool
- `info` - Very short description text about the tool. Supports markdown formatting when `autoformat=True` and `autoformat_type="markdown"` (default). Can include **bold** text, *italic* text, [links](https://example.com), lists, and other markdown features.
- `doi` - One or more publication DOIs (can be a string or a list)
- `comment` - Additional comment text for module. Usually user-supplied in a config.
- `extra` - Optional additional description. Will appear in the documentation and in the report, but not on the list of modules on the website.
- `target` - Name of the module in the description (default: `name`)
- `autoformat` - (default: `True`)
- `autoformat_type` - (default: `markdown`)
:::tip
**Backward Compatibility**: Existing modules with plain text `info` parameters will continue to work as before. The markdown processing only applies when `autoformat=True` (default) and `autoformat_type="markdown"` (default).
:::
Ok, that should be it! The `__init__()` function will now be executed every
time MultiQC runs. Try adding a `print("Hello World!")` statement and see
if it appears in the MultiQC logs at the appropriate time...
### Documentation
If there are any specific considerations for the users before running the module,
add them into the module docstring, e.g.:
````py
from multiqc.base_module import BaseMultiqcModule
class MultiqcModule(BaseMultiqcModule):
"""
The tool provides multiple subcommands, and the MultiQC module currently only
supports `command1`.
The tool outputs useful information into stdout, and you need to capture it to
a file for the module to recognize. To pipe stderr into a file, run the tool
as follows:
```
mymod command1 2> sample1.log
```
Note the that the sample name is parsed from the filename by default, in this case,
the reported name will be "sample1".
#### Configuration
By default, the tool uses the following thresholds to report something: 1, 2, 3.
To override them, use the following config:
```yaml
mymod:
thresholds:
- 1
- 2
- 3
```
Version 1.1.0 of the tool is tested.
"""
def __init__(self):
super().__init__(
...
)
...
The consideration can be:
- The list of supported subcommands of a toolkit;
- The list of supported use cases and sets of parameters;
- Versions of the tools that are supported or tested;
- Required outputs file naming and redirection;
- The way the sample name is found in the logs, if not obvious;
- Configuration parameters that the tool can read from the user config;
- Any post-processing needed to be done by the user before running the module;
- Performance considerations;
- Conflicts with other MultiQC modules.
Logging
Last thing - MultiQC modules have a standardised way of producing output,
so you shouldn't really use print() statements for your Hello World in
your module code ;).
Instead, use the logger module as follows:
import logging
log = logging.getLogger(__name__)
log.info("Hello World!")
Log messages can come in a range of formats:
log.debug- These only show if MultiQC is run in
-v/--verbosemode
- These only show if MultiQC is run in
log.info- For more important status updates
log.warning- Alert user about problems that don't halt execution
log.errorandlog.critical- Not often used, these are for show-stopping problems
Pull-request tags
Pull-request labels/tags are essential for auto-generation of the release changelog, so consider adding them to your PR.
- When opening a pull-request for a new module, please add the
module: newlabel. - If the pull-request only fixes an existing module, please add the
bug: modulelabel. - If it's an enhancement of an existing module, add the
module: enhancementlabel. - If the PR fixes the core codebase, add the
bug: core. - For other options, consider, like
core: frontend,core: refactoring,core: infrastructure(e.g. CI workflows and tests),documentation.
Please do not add anything to the CHANGELOG.md file!
This is now handled by our friendly MultiQC bot 🤖
For more information about how it works, see the contributing docs.
Step 1 - Find log files
The first thing that your module will need to do is to find analysis log files. You can do this by searching for a filename fragment, or a string within the file. It's possible to search for both (a match on either will return the file) and also to have multiple strings possible.
First, add your default patterns to multiqc/search_patterns.yaml
Each search has a yaml key, with one or more search criteria.
The yaml key must begin with the name of your module. If you have multiple
search patterns for a single module, follow the module name with a forward
slash and then any string. For example, see the fastqc module search patterns:
fastqc/data:
fn: "fastqc_data.txt"
fastqc/zip:
fn: "_fastqc.zip"
The following search criteria sub-keys can then be used:
fn- A glob filename pattern, used with the Python
fnmatchfunction
- A glob filename pattern, used with the Python
fn_re- A regex filename pattern
contents- A string to match within the file contents (checked line by line)
contents_re- A regex to match within the file contents (checked line by line)
- NB: Regex must match entire line (add
.*to start and end of pattern to avoid this)
exclude_fn- A glob filename pattern which will exclude a file if matched
exclude_fn_re- A regex filename pattern which will exclude a file if matched
exclude_contents- A string which will exclude the file if matched within the file contents (checked line by line)
exclude_contents_re- A regex which will exclude the file if matched within the file contents (checked line by line)
num_lines- The number of lines to search through for the
contentsstring. Defaults to 1000 (configurable viafilesearch_lines_limit). Set or a low number like 10 if it's e.g. a header of a TSV file. Do not set it to 1 (!!!) because there is a chance that other versions of this file can have extra-headers.
- The number of lines to search through for the
shared- By default, once a file has been assigned to a module it is not searched again. Specify
shared: truewhen your file is likely to be shared between multiple tools, or has a too generic search pattern.
- By default, once a file has been assigned to a module it is not searched again. Specify
max_filesize- Files larger than the
log_filesize_limitconfig key (default: 50MB) are skipped. If you know your files will be smaller than this and need to search by contents, you can specify this value (in bytes) to skip any files smaller than this limit.
- Files larger than the
Please try to use num_lines and max_filesize where possible as they will speed up
MultiQC execution time.
Please do not set num_lines to anything over 1000, as this will significantly slow
down the file search for all users.
If you do need to search more lines to detect a string, please combine it with
a fn pattern to limit which files are loaded (as done with AfterQC).
For example, two typical modules could specify search patterns as follows:
mymodule:
fn: "_myprogram.txt"
myother_module:
contents: "This is myprogram v1.3"
You can also supply a list of different patterns for a single log file type if needed. If any of the patterns are matched, the file will be returned:
mymodule:
- fn: "mylog.txt"
- fn: "different_fn.out"
You can use AND logic by specifying keys within a single list item. For example:
mymodule:
fn: "mylog.txt"
contents: "mystring"
myother_module:
- fn: "different_fn.out"
contents: "This is myprogram v1.3"
- fn: "another.txt"
contents: ["What are these files anyway?", "End of program"]
contents_re: '^Metric: \d+\.\d+'
For mymodule, a file must have the filename mylog.txt and contain the string mystring.
myother_module will match different_fn.out with the contents This is myprogram v1.3,
or another.txt containing ALL of the lines What are these files anyway?, End of program",
and ^Metric: \d+\.\d+.
You can match subsets of files by using exclude_ keys as follows:
mymodule:
fn: "*.myprog.txt"
exclude_fn: "not_these_*"
myother_module:
fn: "mylog.txt"
exclude_contents:
- "trimmed"
- "sorted"
Note that the exclude_ patterns can have either a single value or a list of values.
They are always considered using OR logic - any matches will reject the file.
Remember that users can overwrite these defaults in their own config files. This is helpful as people have weird and wonderful processing pipelines with their own conventions.
Once your strings are added, you can find files in your module with the
base function self.find_log_files(), using the key you set in the YAML:
self.find_log_files("mymodule")
This function yields a dictionary with various information about each matching
file. The f key contains the contents of the matching file:
# Find all files for mymod
for f in self.find_log_files("mymodule"):
print(f["f"]) # File contents
print(f["s_name"]) # Sample name (from cleaned filename)
print(f["fn"]) # Filename
print(f["root"]) # Directory file was in
If filehandles=True is specified, the f key contains a file handle
instead:
for f in self.find_log_files("mymodule", filehandles=True):
# f['f'] is now a filehandle instead of contents
for line in f["f"]:
print(line)
This is good if the file is large, as Python doesn't read the entire file into memory in one go.
Step 2 - Parse data from the input files
What most MultiQC modules do once they have found matching analysis files is to pass the matched file contents to another function, responsible for parsing the data from the file. How this parsing is done will depend on the format of the log file and the type of data being read. See below for a basic example, based loosely on the preseq module:
from multiqc.base_module import BaseMultiqcModule
from typing import Dict, Union
class MultiqcModule(BaseMultiqcModule):
def __init__(self):
...
data_by_sample: Dict[str, Dict[str, Union[float, int]]] = dict()
for f in self.find_log_files("mymod"):
s_name = f["s_name"]
if s_name in data_by_sample:
log.debug(f"Duplicate sample name found! Overwriting: {s_name}")
data_by_sample[s_name] = parse_file(f["f"])
def parse_file(f) -> Dict[str, Union[float, int]]:
data = {}
for line in f.splitlines():
s = line.strip().split()
data[s[0]] = float(s[1])
return data
Filtering by parsed sample names
MultiQC users can use the --ignore-samples flag to skip sample names
that match specific patterns. As sample names are generated in a different
way by every module, this filter has to be applied after log parsing.
There is a core function to do this task - assuming that your data is
in a dictionary with the first key as sample name, pass it through the
self.ignore_samples function as follows:
data_by_sample = ...
data_by_sample = self.ignore_samples(data_by_sample)
This will remove any dictionary keys where the sample name matches a user pattern.
If your data structure is not in the sample_name: data format then
you can check each sample name individually using the
self.is_ignore_sample() function:
if self.is_ignore_sample(f["s_name"]):
print("We will not use this sample!")
Note that this function should be used after cleaning the sample name with
self.clean_s_name().
No files found
If your module cannot find any matching files, it needs to raise an
exception of type ModuleNoSamplesFound. This tells the core MultiQC program
that no modules were found. For example:
from multiqc.base_module import ModuleNoSamplesFound
if len(data_by_sample) == 0:
raise ModuleNoSamplesFound
Note that this has to be raised as early as possible, so that it halts the module progress. For example, if no logs are found then the module should not create any files or try to do any computation.
Custom sample names
Typically, sample names are taken from cleaned log filenames (the default
f['s_name'] value returned). However, if the underlying tool records the sample
name in the logs somewhere, it's better to use that instead. Alternatively, it could
also record the name of the input file somewhere (e.g. adapter cleaning tools typically
save the input FASTQ file name in the log), in which case it's better to clean the
sample name from the input file name. For that, you should use the self.clean_s_name()
method, as this will prepend the directory name if requested on the command line:
for f in self.find_log_files("mymodule"):
input_fname, data = parse_file(f)
s_name = self.clean_s_name(input_fname, f)
...
This function has already been applied to the contents of f['s_name'],
so it is only required when using something different for the sample identifier.
self.clean_s_name() must be used on sample names parsed from the file
contents. Without it, features such as prepending directories (--dirs)
will not work.
The second argument should be the dictionary returned by the self.find_log_files() function.
The root path is used for --dirs and the search pattern key is used
for fine-grained configuration of the config option use_filename_as_sample_name.
If you are using non-standard values for the logfile root, filename or search pattern key, these can be specified. The function def looks like this:
def clean_s_name(self, s_name, f, root=None):
A typical example is when the sample name is the log file directory. In this case, the root should be the dirname of that directory. This is non-standard, and would be specified as follows:
s_name = self.clean_s_name(f["root"], f, root=os.path.dirname(f["root"]))
Identical sample names
If modules find samples with identical names, then the previous sample
is overwritten. It's good to print a log statement when this happens,
for debugging. However, most of the time it makes sense - programs often
create log files and print to stdout for example.
if f["s_name"] in data_by_sample:
log.debug(f"Duplicate sample name found! Overwriting: {f['s_name']}")
Printing to the sources file
Finally, once you've found your file we want to add this information to the
multiqc_sources.txt file in the MultiQC report data directory. This lists
every sample name and the file from which this data came from. This is especially
useful if sample names are being overwritten as it lists the source used. This code
is typically written immediately after the above warning.
If you've used the self.find_log_files function, writing to the sources file
is as simple as passing the log file variable to the self.add_data_source function:
for f in self.find_log_files("mymodule"):
self.add_data_source(f)
If you have different files for different sections of the module, or are customising the sample name, you can tweak the fields. The default arguments are as shown:
self.add_data_source(f=None, s_name=None, source=None, module=None, section=None)
Saving version information
Software version information may be present in the log files of some tools. The
version number can be included in the report by passing it to the method
self.add_software_version. Let's use this samtools stats log below as an example.
# This file was produced by samtools stats (1.3+htslib-1.3) and can be plotted using plot-bamstats
# This file contains statistics for all reads.
# The command line was: stats /home/lp113/bcbio-nextgen/tests/test_automated_output/align/Test1/Test1.sorted.bam
# CHK, Checksum [2]Read Names [3]Sequences [4]Qualities
# CHK, CRC32 of reads which passed filtering followed by addition (32bit overflow)
CHK 560674ab 1165a6ca 7b309ac6
# Summary Numbers. Use `grep ^SN | cut -f 2-` to extract this part.
SN raw total sequences: 101
...
The version number here (1.3) can be extracted using a regular expression (regex).
We then pass this to the self.add_software_version() function.
Note that we pass the sample name (f["s_name"] in this case) so that we don't
add versions for samples that are later ignored.
import re
for line in f.splitlines():
version = re.search(r"# This file was produced by samtools stats \(([\d\.]+)", line)
if version is not None:
self.add_software_version(version.group(1), sample=f["s_name"])
# ..rest of file parsing
The version number will now appear after the module header in the report as well as in the section Software Versions in the end of the report.
For tools that don't output software versions in their logs these can instead be provided in a separate YAML file. See Customising Reports for details.
In some cases, a log may include multiple version numbers for a single tool.
In the example provided, the version of htslib is shown alongside the
previously extracted samtools version. This information is valuable and
should be incorporated into the report. To achieve this, we need to
extract the new version string and provide it to the
self.add_software_version() function. Include the relevant software
name (in this case, htslib) as well. This will ensure that the htslib
version is listed separately from the main module's software version.
Example:
for line in f.splitlines():
version = re.search(r"# This file was produced by samtools stats \(([\d\.]+)", line)
if version is not None:
self.add_software_version(version.group(1), sample=f["s_name"])
htslib_version = re.search(r"\+htslib-([\d\.]+)", line)
if htslib_version is not None:
self.add_software_version(htslib_version.group(1), sample=f["s_name"], software_name="htslib")
... # rest of file parsing
Even if the logs does not contain any version information, you should still
add a superfluous self.add_software_version() call to the module. This
will help maintainers to check if new modules or submodules parse any version
information that might exist. The call should also include a note that it is
a dummy call. Example:
for f in self.find_log_files("mymodule/submodule"):
sample = f["s_name"]
data_by_sample[sample] = parse_file(f)
# Superfluous function call to confirm that it is used in this module
# Replace None with actual version if it is available
self.add_software_version(None, sample)
Step 3 - Adding to the general statistics table
Now that you have your parsed data, you can start inserting it into the MultiQC report. At the top of every report is the 'General Statistics' table. This contains metrics from all modules, allowing cross-module comparison.
Do not add a lot of columns to the General Statistics table. There should be 1-2 columns visible-by-default columns per module, plus there can be a bunch of more hidden columns.
There is a helper function to add your data to this table. It can take a lot of configuration options, but most have sensible defaults. At it's simplest, it works as follows:
data_by_sample: Dict[str, Dict[str, float]] = {
"sample_1": {
"first_col": 91.4,
"second_col": 78.2,
},
"sample_2": {
"first_col": 138.3,
"second_col": 66.3,
},
}
self.general_stats_addcols(data_by_sample)
To give more informative table headers and configure things like data scales and colour schemes, you can supply an extra dict:
from multiqc.plots.table_object import ColumnMeta
headers = {
"first_col": ColumnMeta(
title="First",
description="My First Column",
scale="RdYlGn-rev",
),
"second_col": ColumnMeta(
title="Second",
description="My Second Column",
max=100,
min=0,
scale="Blues",
suffix="%",
)
}
self.general_stats_addcols(data_by_sample, headers)
Here are all options for headers, with defaults:
headers["name"] = TableColumn(
namespace="", # Module name. Auto-generated for core modules in General Statistics.
title="[ dict key ]", # Short title, table column title
description="[ dict key ]", # Longer description, goes in mouse hover text
max=None, # Minimum value in range, for bar / colour coding
min=None, # Maximum value in range, for bar / colour coding
scale="GnBu", # Colour scale for colour coding. Set to False to disable.
suffix=None, # Suffix for value (eg. '%')
format="{:,.1f}", # Output format() string. Can also be a lambda function.
shared_key=None, # See below for description
modify=None, # Lambda function to modify values
hidden=False, # Set to True to hide the column on page load
placement=1000.0, # Alter the default ordering of columns in the table
)
namespace- This prepends the column title in the mouse hover: Namespace: Title.
- The 'Configure Columns' modal displays this under the 'Group' column.
- It's automatically generated for core modules in the General Statistics table, though this can be overwritten (useful for example with custom-content).
scale- Colour scales are the names of ColorBrewer palettes. See below for available scales.
- Add
-revto the name of a colour scale to reverse it - Set to
Falseto disable colouring and background bars
shared_key- Any string can be specified here, if other columns are found that share
the same key, a consistent colour scheme and data scale will be used in
the table. Typically this is set to things like
read_count, so that the read count in a sample can be seen varying across analysis modules.
- Any string can be specified here, if other columns are found that share
the same key, a consistent colour scheme and data scale will be used in
the table. Typically this is set to things like
modify- A python
lambdafunction to change the data in some way when it is inserted into the table.
- A python
format- A format string or a python
lambdafunction to format the data to display on screen.
- A format string or a python
hidden- Setting this to
Truewill hide the column when the report loads. It can then be shown through the Configure Columns modal in the report. This can be useful when data could be sometimes useful. For example, some modules show "percentage aligned" on page load but hide "number of reads aligned".
- Setting this to
placement- If you feel that the results from your module should appear on the left side of the table set this value less than 1000. Or to move the column right, set it greater than 1000. This value can be any float.
The typical use for the modify string is to divide large numbers such as read counts,
to make them easier to interpret. If handling read counts, there are three config variables
that should be used to allow users to change the multiplier for read counts:
read_count_multiplier, read_count_prefix and read_count_desc. For example:
from multiqc.plots.table_object import TableConfig
pconfig = TableConfig(
title="Reads",
description=f"Number of reads ({config.read_count_desc})",
modify=lambda x: x * config.read_count_multiplier,
suffix=f" {config.read_count_prefix}",
...
)
Similar config options apply for base pairs: base_count_multiplier, base_count_prefix and
base_count_desc.
And for the read count of long reads: long_read_count_multiplier, long_read_count_prefix and
long_read_count_desc.
Note that adding e.g. "shared_key": "read_count" will automatically add corresponding
description, modify, and suffix into the column, so in most cases the following
will be sufficient:
pconfig = TableConfig(
title="Reads",
shared_key="read_count",
...
)
...
pconfig2 = TableConfig(
title="Base pairs",
shared_key="base_count",
...
)
A third parameter can be passed to this function, namespace. This is usually
not needed - MultiQC automatically takes the name of the module that is calling
the function and uses this. However, sometimes it can be useful to overwrite this.
Table colour scales
Colour scales are taken from ColorBrewer2.
Colour scales can be reversed by adding the suffix -rev to the name. For example, RdYlGn-rev.
The following scales are available:

For categorical metrics that can take a value from a predefined set, use one of the categorical color scales: Set2, Accent, Set1, Set3, Dark2, Paired, Pastel2, Pastel1. For numerical metrics, consider one the "sequential" color scales from the table above.
Grouping samples
If you have a set of samples that should be grouped together in the report using the sample grouping configuration option, you can include the group_samples_config parameter to the self.general_stats_addcols function. For example, FastQC uses the following configuration:
self.general_stats_addcols(
...,
group_samples_config=SampleGroupingConfig(
cols_to_sum=[ColumnKey("total_sequences")],
cols_to_weighted_average=[
(ColumnKey("percent_gc"), ColumnKey("total_sequences")),
(ColumnKey("avg_sequence_length"), ColumnKey("total_sequences")),
(ColumnKey("percent_duplicates"), ColumnKey("total_sequences")),
(ColumnKey("median_sequence_length"), ColumnKey("total_sequences")),
],
extra_functions=[_summarize_statues],
)
)
In this configuration, you can specify how to merge data for each column:
cols_to_sum- add up values for each sample in the group.cols_to_average- take an average of all samples.cols_to_weighted_average- take a weighed average, specifying the weight column in the the second tuple parameter.extra_functions- list of functions to call to add extra data to the merged rowexplicit_groups- opt-in dict of{group_display_name: [sample_names]}that lets a module supply its own ground-truth groups instead of relying on the user-suppliedtable_sample_mergename patterns.
Extra functions
The extra_functions flag can be used when you need custom logic beyond summing or averaging numeric values. For example, FastQC uses it to recalculate the percent_fails value:
def _summarize_statues(
merged_row: InputRow, group_s_names: List[Tuple[Optional[str], SampleName, SampleName]]
):
# Add count of fail statuses
_num_statuses = 0
_num_fails = 0
for _, _, original_sn in group_s_names:
for st in self.fastqc_data[original_sn]["statuses"].values():
_num_statuses += 1
if st == "fail":
_num_fails += 1
if _num_statuses > 0:
merged_row.data[ColumnKey("percent_fails")] = (float(_num_fails) / float(_num_statuses)) * 100.0
Explicit groups
Use this when the tool output already tells you which samples are related. For example a tool whose log carries a stable pair / replicate identifier. When set, this short-circuits the name-pattern matcher: each [sample_names] list is collapsed into one group with the given display name.
# Build the groups from tool metadata during parse.
explicit_groups: Dict[str, List[str]] = {}
for s_name, data in data_by_sample.items():
group_id = data["sample_id"] # This will depend on your data structure
explicit_groups.setdefault(group_id, []).append(s_name)
self.general_stats_addcols(
data_by_sample,
headers,
group_samples_config = SampleGroupingConfig(
explicit_groups = explicit_groups,
cols_to_sum = [ColumnKey("some_count_column")],
cols_to_weighted_average = [
(ColumnKey("some_percent"), ColumnKey("some_count_column"))
],
),
)
Entries with a single member are ignored by the framework - they fall through and render as a normal ungrouped row with their original sample name.
Modules that use this should expose a config flag so users can opt out and see per-sample rows.
Auto-grouping is independent of table_sample_merge: if the user has also configured name patterns, you can layer them on top of your auto-groups before passing: run each auto-group's display name through self.groups_for_sample(...) and bucket by the result.
Step 4 - Writing data to a file
In addition to printing data to the General Stats, MultiQC modules typically also write to text-files to allow people to easily use the data in downstream applications. This also gives the opportunity to output additional data that may not be appropriate for the General Statistics table.
Again, there is a base class function to help you with this - just supply it with a dictionary and a filename:
data_by_sample = {
"sample_1": {
"first_col": 91.4,
"second_col": "78.2%",
},
"sample_2": {
"first_col": 138.3,
"second_col": "66.3%",
},
}
self.write_data_file(data_by_sample, "multiqc_mymodule")
Make sure to call self.write_data_file in the end of the module, because it
may modify data_by_sample to be JSON-serializable.
If your output has a lot of columns, you can supply the additional
argument sort_cols = True to have the columns alphabetically sorted.
This function will also pay attention to the default / command line
supplied data format and behave accordingly. So the written file could
be a tab-separated file (default), JSON or YAML.
Note that any keys with more than 2 levels of nesting will be ignored when being written to tab-separated files.
Step 5 - Create report sections
Great! It's time to start creating sections of the report with more information.
To do this, use the self.add_section() helper function.
This supports the following arguments:
name: Name of the section, used for the titleanchor: The URL anchor - must be unique, used when clicking the name in the side-navdescription: A very short descriptive text to go above the plot (markdown).comment: A comment to add under the description. Big and blue text, mostly for users to customise the report (markdown).helptext: Longer help text explaining what users should look for (markdown).plot: Results from one of the MultiQC plotting functionscontent: Any custom HTMLautoformat: DefaultTrue. Automatically format thedescription,commentandhelptextstrings.autoformat_type: Defaultmarkdown. Autoformat text type. Currently onlymarkdownsupported.statuses: Optional dictionary with keys"pass","warn", and"fail", each containing lists of sample names. When provided, adds an interactive status progress bar to the section header showing pass/warn/fail counts.alerts: Optional alert box, or list of alert boxes, shown below the description. Alert messages support markdown, Bootstrap alert levels, and optional affected sample lists.
Section status bars
If your tool generates pass/warn/fail metrics for different QC checks, you can add interactive status bars to section headers using the statuses parameter in add_section().
The status bars will automatically:
- Show sample lists on hover (after 0.5s delay)
- Pin the popover on click
- Provide "Highlight" and "Filter" buttons for integration with the MultiQC toolbox
- Display colored progress bars showing the proportion of samples in each status category
For example:
# Collect sample names by status for this section
status_data = {
"pass": ["sample1", "sample2", "sample3"],
"warn": ["sample4"],
"fail": ["sample5"]
}
# Add section with status bar
self.add_section(
name="Quality Check",
anchor="quality_check",
description="Results from quality control analysis",
plot=my_plot,
statuses=status_data
)
Status bar user configuration
Users can control status bar visibility globally or per-module using the section_status_checks config:
section_status_checks:
fastqc: false # Disable all FastQC status bars
mymodule:
section1: false # Disable specific section status bar
By default, all status bars are enabled. Configuration can be set at the module level (boolean) or per-section level (nested dictionary).
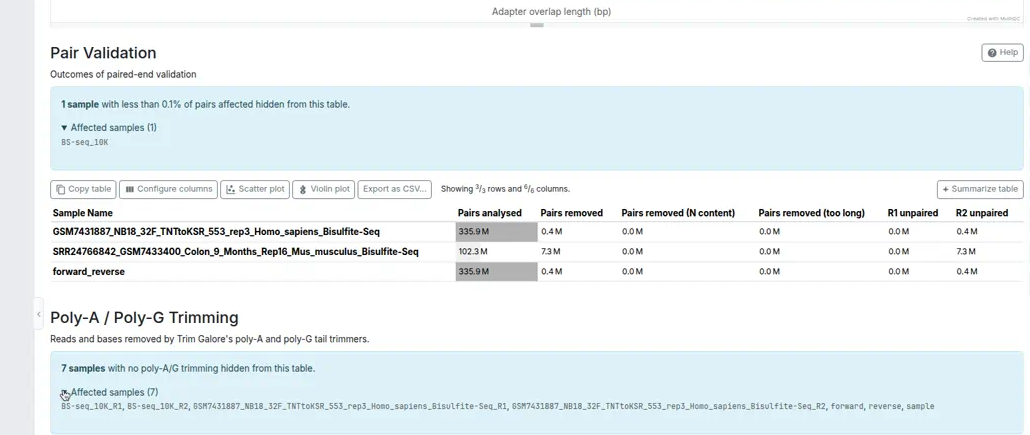
Section alerts
Use the alerts parameter when a section needs to call out an important note, especially if samples were hidden from a plot or table.
This keeps warnings separate from the section description and lets MultiQC render the Bootstrap alert markup consistently.
Each alert can be a plain markdown string, a dictionary, a SectionAlert, or a list of these.
Dictionary and SectionAlert values support:
message: Alert text, formatted as markdown by defaultlevel: Bootstrap alert style, default"info"; must be one of"primary","secondary","success","danger","warning","info","light", or"dark"affected_samples: Optional list of sample names, rendered in an expandable list
Alerts with an empty message are ignored.
After add_section() runs, the SectionAlert.message value stored on the section is the rendered HTML string, matching how section descriptions are stored.
For example:
from multiqc.types import SectionAlert
self.add_section(
name="Adapter Content",
anchor="adapter_content",
description="Adapter content per cycle.",
plot=adapter_plot,
alerts=SectionAlert(
message="**3 samples** with negligible adapter content hidden from this plot.",
level="warning",
affected_samples=["sample1", "sample2", "sample3"],
),
)
Sections with alerts still render even when there is no plot or custom content.
Use alerts instead of appending raw <div class="alert ..."> HTML to description or content.
For example:
from multiqc.plots import linegraph, bargraph
from multiqc.plots.linegraph import LinePlotConfig
from multiqc.plots.bargraph import BarPlotConfig
self.add_section(
name="Second Module Section",
anchor="mymodule-second",
plot=linegraph.plot(data_by_sample2, pconfig=LinePlotConfig(
id="mymodule-second",
title="My Module: Duplication Rate"
)),
)
self.add_section(
name="First Module Section",
anchor="mymodule-first",
description="My amazing module output, from the first section",
helptext="""
If you're not sure _how_ to interpret the data, we can help!
Most modules use multi-line strings for these text blocks,
with triple quotation marks.
* Markdown
* Lists
* Are
* `Great`
""",
plot = bargraph.plot(data_by_sample, pconfig=BarPlotConfig(
id="mymodule-first",
title="My Module: Read Counts"
))
)
self.add_section(
content="<p>Some custom HTML.</p>"
)
If a module has more than one section, these will automatically be labelled and linked
in the left sidebar navigation (unless name is not specified).
Step 6 - Plot some data
Ok, you have some data, now the fun bit - visualising it! Each of the plot types is described in the Plotting Functions section of the docs.
Appendices
User configuration
Instead of hard-coding the defaults, it's a great idea to allow users to configure the behaviour of MultiQC module code.
It's pretty easy to use the built-in MultiQC configuration settings to do this, so that users can set up their config as described in the Configuration docs.
To do this, just assume that your configuration variables are available in the
MultiQC config module and have sensible defaults. For example:
from multiqc import config
mymod_config = getattr(config, 'mymod', {})
my_custom_config_var = mymod_config.get('my_custom_config_var', 5)
You now have a variable my_custom_config_var with a default value of 5, but that
can be configured by a user as follows:
mymod:
my_custom_config_var: 200
Please be sure to use a unique top-level config name to avoid clashes - prefixing with your module name is a good idea as in the example above. Keep all module config options under the same top-level name for clarity.
Finally, don't forget to document the usage of your module-specific configuration
in the MultiqcModule class docstring, so that people know how to use it.
Profiling Performance
It's important that MultiQC runs quickly and efficiently, especially on big
projects with large numbers of samples. The recommended method to check this is
by using cProfile to profile the code execution.
To do this, first find out where your copy of MultiQC is located:
$ which multiqc
/Users/you/anaconda/envs/myenv/bin/multiqc
Then run MultiQC with this path and the cProfile module as follows
(the flags at the end can be any regular MultiQC flags):
python -m cProfile -o multiqc_profile.prof /Users/you/anaconda/envs/myenv/bin/multiqc -f .
You can create a .bashrc alias to make this easier to run:
alias profile_multiqc='python -m cProfile -o multiqc_profile.prof /Users/you/anaconda/envs/myenv/bin/multiqc '
profile_multiqc -f .
MultiQC should run as normal, but produce the additional binary file multiqc_profile.prof.
This can then be visualised with software such as SnakeViz.
To install SnakeViz and visualise the results, do the following:
pip install snakeviz
snakeviz multiqc_profile.prof
A web page should open where you can explore the execution times of different nested functions. It's a good idea to run MultiQC with a comparable number of results from other tools (eg. FastQC) to have a reference to compare against for how long the code should take to run.
Adding Custom CSS / Javascript
If you would like module-specific CSS and / or JavaScript added to the template,
just add to the self.css and self.js dictionaries that come with the
BaseMultiqcModule class. The key should be the filename that you want your file to
have in the generated report folder (this is ignored in the default template, which
includes the content file directly in the HTML). The dictionary value should be
the path to the desired file. For example, see how it's done in the FastQC module:
import os
self.css = {
"assets/css/multiqc_fastqc.css": os.path.join(os.path.dirname(__file__), "assets", "css", "multiqc_fastqc.css")
}
self.js = {
"assets/js/multiqc_fastqc.js": os.path.join(os.path.dirname(__file__), "assets", "js", "multiqc_fastqc.js")
}
Addendum - example module
Below is an example of a good-quality module written for a made-up tool Qualalyser:
File system structure:
├── multiqc
│ ├── modules
│ | └── qualalyser
│ │ ├── __init__.py
│ │ ├── qualalyser.py
│ │ └── tests
│ │ ├── __init__.py
│ │ └── test_qualalyser.py
│ └── search_patterns.yaml
└── pyproject.toml
__init__.py
from .qualalyser import MultiqcModule
__all__ = ["MultiqcModule"]
qualalyser.py
import logging
import re
from collections import defaultdict
from copy import deepcopy
from typing import Callable, Dict, List, Any, Tuple, Union
from multiqc.base_module import BaseMultiqcModule, ModuleNoSamplesFound
from multiqc.plots import table, bargraph
from multiqc.plots.bargraph import BarPlotConfig
from multiqc.plots.table_object import TableConfig, ColumnMeta
from multiqc.utils import mqc_colour
from multiqc import config
log = logging.getLogger(__name__)
class MultiqcModule(BaseMultiqcModule):
"""
Qualalyser provides multiple subcommands, and the MultiQC module currently only supports `quality`.
Qualalyser outputs useful information into stdout, and you need to capture it to
a file for the module to recognize. To pipe stderr into a file, run the tool
as follows:
qualalyser quality 2> sample1.log
Note the that the sample name is parsed from the filename by default, in this case,
the reported name will be "sample1".
#### Configuration
By default, Qualalyser uses the following quality threshold: 10.
To override it, use the following config:
qualalyser:
min_quality: 10
Version 1.1.0 of Qualalyser is tested.
"""
def __init__(self):
super().__init__(
name="Qualalyser",
anchor="qualalyser",
href="https://github.com/bioinformatics-centre/qualalyser/",
info="Reports read quality and length from sequencing data",
doi="10.21105/joss.02991",
)
# Find and load any Qualalyser reports
data_by_sample: Dict[str, Dict[str, float]] = {}
for f in self.find_log_files("qualalyser/quality", filehandles=True):
sample_data = parse_qualalyser_log(f)
if sample_data:
s_name = f['s_name']
if s_name in data_by_sample:
log.debug(f"Duplicate sample name found! Overwriting: {s_name}")
data_by_sample[s_name] = sample_data
self.add_data_source(f)
# Superfluous function call to confirm that it is used in this module
# Replace None with actual version if it is available
self.add_software_version(None)
# Filter to strip out ignored sample names
data_by_sample = self.ignore_samples(data_by_sample)
if len(data_by_sample) == 0:
raise ModuleNoSamplesFound
log.info(f"Found {len(data_by_sample)} reports")
# Add Qualalyser summary to the general stats table
self.add_table(data_by_sample)
# Quality distribution Plot
self.reads_by_quality_plot(data_by_sample)
# Read length distribution Plot
self.reads_by_length_plot(data_by_sample)
# Write parsed report data to a file
self.write_data_file(data_by_sample, "multiqc_qualalyser")
def add_table(self, data_by_sample: Dict[str, Dict[str, float]]) -> None:
headers: Dict[str, Dict] = {
"Number of reads": ColumnMeta(
title="Reads",
description="Number of reads",
scale="Greens",
shared_key="read_count",
),
"Number of bases": ColumnMeta(
title="Bases",
description="Total bases sequenced",
scale="Purples",
shared_key="base_count",
),
"N50 read length": ColumnMeta(
title="Read N50",
description="N50 read length",
scale="Blues",
suffix="bp",
format="{:,.0f}",
),
"Longest read": ColumnMeta(
title="Longest Read",
description="Longest read length",
suffix="bp",
scale="Oranges",
format="{:,.0f}",
),
"Mean read length": ColumnMeta(
title="Mean Length",
description="Mean read length",
suffix="bp",
scale="PuBuGn",
),
"Median read length": ColumnMeta(
title="Median Length",
description="Median read length (bp)",
scale="RdYlBu",
format="{:,.0f}",
),
"Mean read quality": ColumnMeta(
title="Mean Qual",
description="Mean read quality (Phred scale)",
scale="PiYG",
),
"Median read quality": ColumnMeta(
title="Median Qual",
description="Median read quality (Phred scale)",
scale="Spectral",
),
}
self.add_section(
name="Qualalyser Summary",
anchor="qualalyser-summary",
description="Statistics from Qualalyser reports",
plot=table.plot(
data_by_sample,
headers,
pconfig=TableConfig(
id="qualalyser_table",
title="Qualalyser Summary",
),
),
)
# Add general stats table - hide all columns except for two
general_stats_headers = deepcopy(headers)
for h in general_stats_headers.values():
h["hidden"] = True
general_stats_headers["Number of reads"]["hidden"] = False
general_stats_headers["N50 read length"]["hidden"] = False
# Add columns to the general stats table
self.general_stats_addcols(data_by_sample, general_stats_headers)
def reads_by_quality_plot(self, data_by_sample: Dict[str, Dict[str, float]]) -> None:
barplot_data: Dict[str, Dict[str, float]] = defaultdict(dict)
keys: List[str] = []
min_quality = getattr(config, "qualalyser", {}).get("min_quality", 10)
for name, d in data_by_sample.items():
reads_by_q = {int(re.search(r"\d+", k).group(0)): v for k, v in d.items() if k.startswith("Reads > Q")}
if not reads_by_q:
continue
thresholds = sorted(th for th in reads_by_q if th >= min_quality)
if not thresholds:
continue
barplot_data[name], keys = get_ranges_from_cumsum(
data=reads_by_q, thresholds=thresholds, total=d["Number of reads"], formatter=lambda x: f"Q{x}"
)
colours = mqc_colour.mqc_colour_scale("RdYlGn-rev", 0, len(keys))
cats = {
k: {"name": f"Reads {k}", "color": colours.get_colour(idx, lighten=1)} for idx, k in enumerate(keys[::-1])
}
# Plot
self.add_section(
name="Read quality",
anchor="qualalyser_plot_quality",
description="Read counts categorised by read quality (Phred score).",
helptext="""
Sequencing machines assign each generated read a quality score using the
[Phred scale](https://en.wikipedia.org/wiki/Phred_quality_score).
The phred score represents the liklelyhood that a given read contains errors.
High quality reads have a high score.
""",
plot=bargraph.plot(
barplot_data,
cats,
pconfig=bargraph.BarPlotConfig(
id="qualalyser_plot_quality_plot",
title="Qualalyser: read qualities",
),
),
)
def parse_qualalyser_log(f) -> Dict[str, float]:
"""Parse output from Qualalyser"""
stats: Dict[str, float] = dict()
# Parse the file content
segment = None
summary_lines = []
length_threshold_lines = []
quality_threshold_lines = []
for line in f["f"]:
line = line.strip()
if line.startswith("Qualalyser Read Summary"):
segment = "summary"
continue
elif line.startswith("Read length thresholds"):
segment = "length_thresholds"
continue
elif line.startswith("Read quality thresholds"):
segment = "quality_thresholds"
continue
if segment == "summary":
summary_lines.append(line)
elif segment == "length_thresholds":
length_threshold_lines.append(line)
elif segment == "quality_thresholds":
quality_threshold_lines.append(line)
for line in summary_lines:
if ":" in line:
metric, value = line.split(":", 1)
stats[metric.strip()] = float(value.strip())
return stats
pyproject.toml
...
[project.entry-points."multiqc.modules.v1"]
qualalyser = "multiqc.modules.qualalyser:MultiqcModule"
...
search_patterns.yaml
---
qualalyser/quality:
fn: "*.log"
contents: "Qualalyser Read Summary"
num_lines: 10
config_defaults.yaml
---
# Order that modules should appear in report. Try to list in order of analysis.
module_order: ...
- qualalyser
...