Community Showcase
The Community Showcase is a Seqera-managed demonstration workspace with everything you need to follow this tutorial. All Seqera Cloud users can access it by default.
This tutorial shows you how to:
- Launch, monitor, and optimize the nf-core/rnaseq pipeline.
- Select pipeline input data with Data Explorer and Platform datasets.
- Analyze pipeline results interactively with Studios.
Use the Launchpad in any workspace to create and share Nextflow pipelines that run on any supported infrastructure, including all public clouds and most high-performance computing (HPC) schedulers. A Launchpad pipeline consists of a pre-configured pipeline repository, compute environment, and launch parameters.
The Community Showcase contains 15 preconfigured pipelines, including nf-core/rnaseq, a bioinformatics pipeline used to analyze RNA sequencing data.
The workspace also includes three preconfigured AWS Batch compute environments to run Community Showcase pipelines, plus Platform datasets and public data sources (accessed through Data Explorer) for use as pipeline input.
To skip this Community Showcase demo and start running pipelines on your own infrastructure:
- Set up an organization workspace.
- Create a workspace compute environment for your cloud or HPC compute infrastructure.
- Add pipelines to your workspace.
Launch the nf-core/rnaseq pipeline
This guide is based on version 3.14.0 of the nf-core/rnaseq pipeline. Launch form parameters may differ in other versions.
Go to the Launchpad in the community/showcase workspace and select Launch next to the nf-core-rnaseq pipeline to open the launch form.
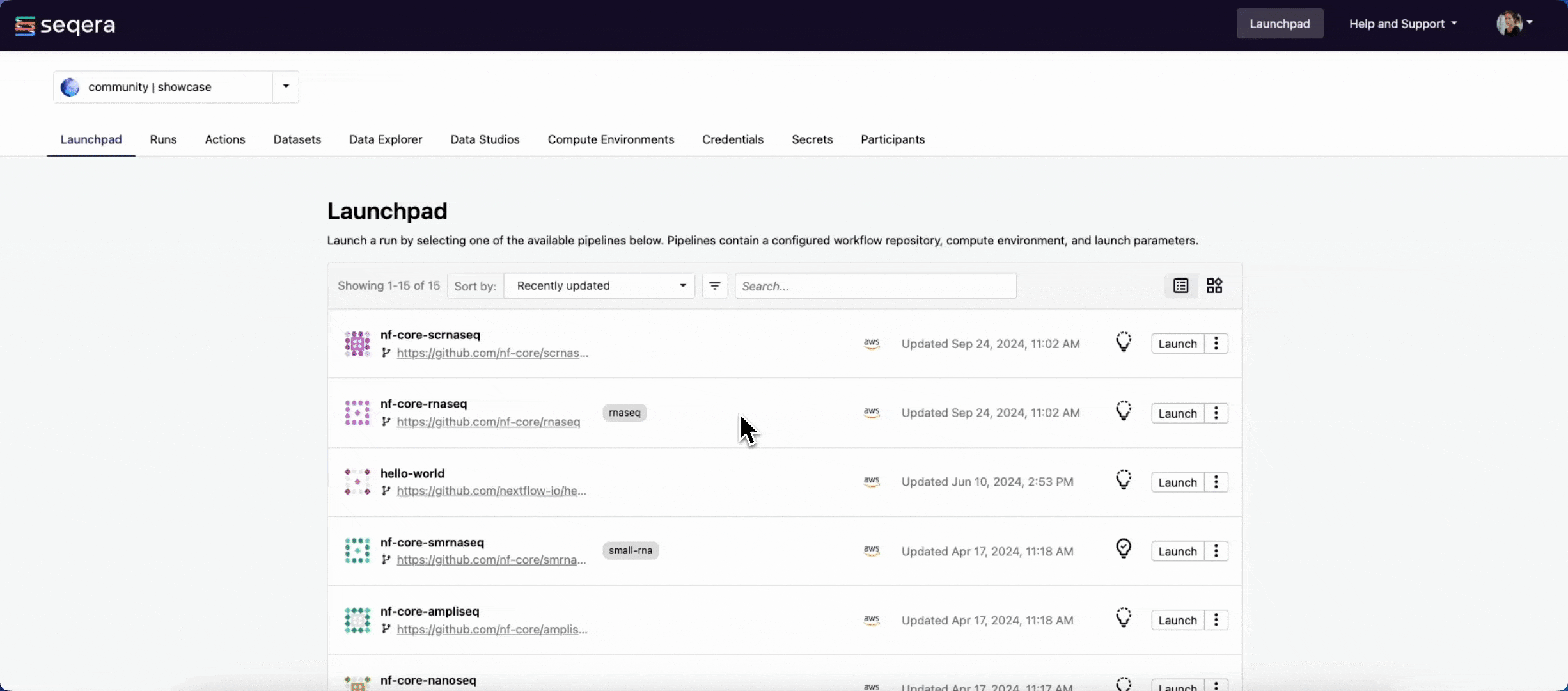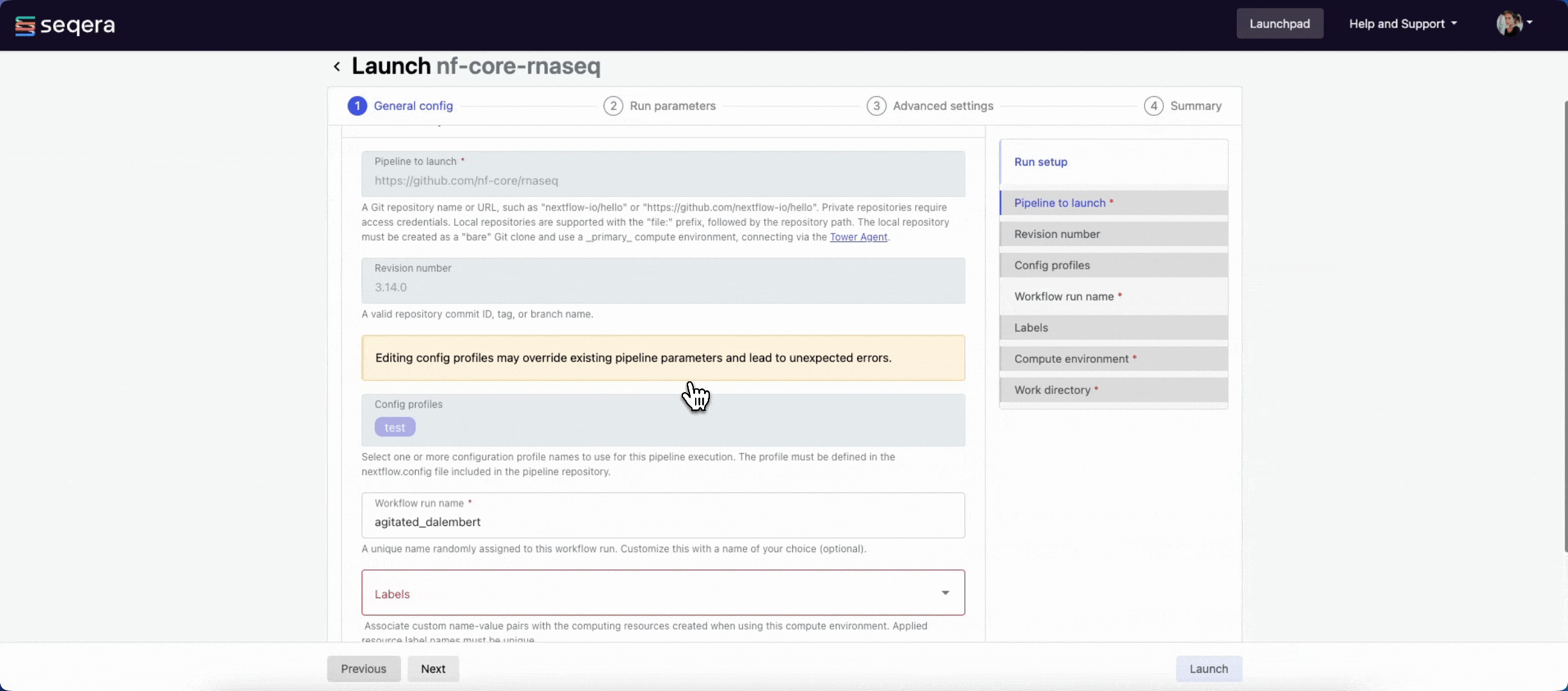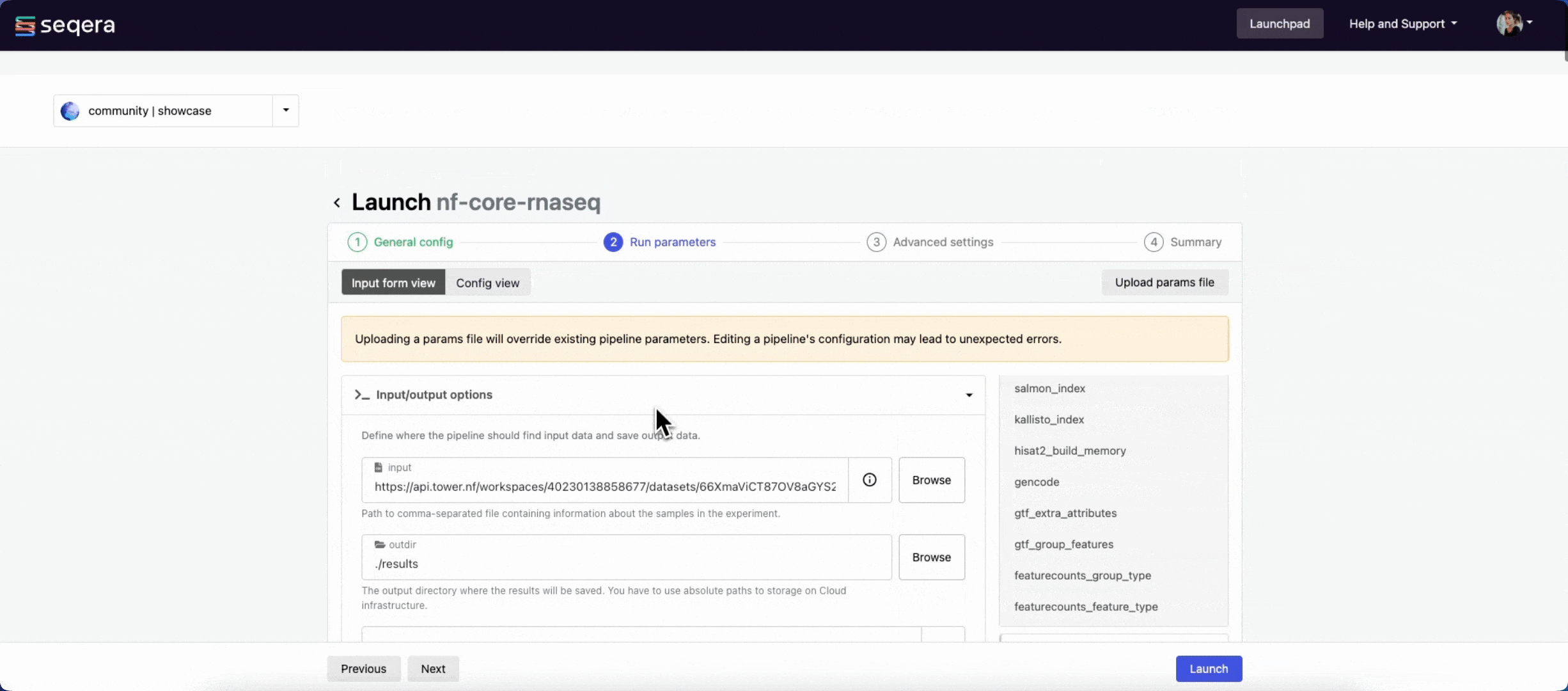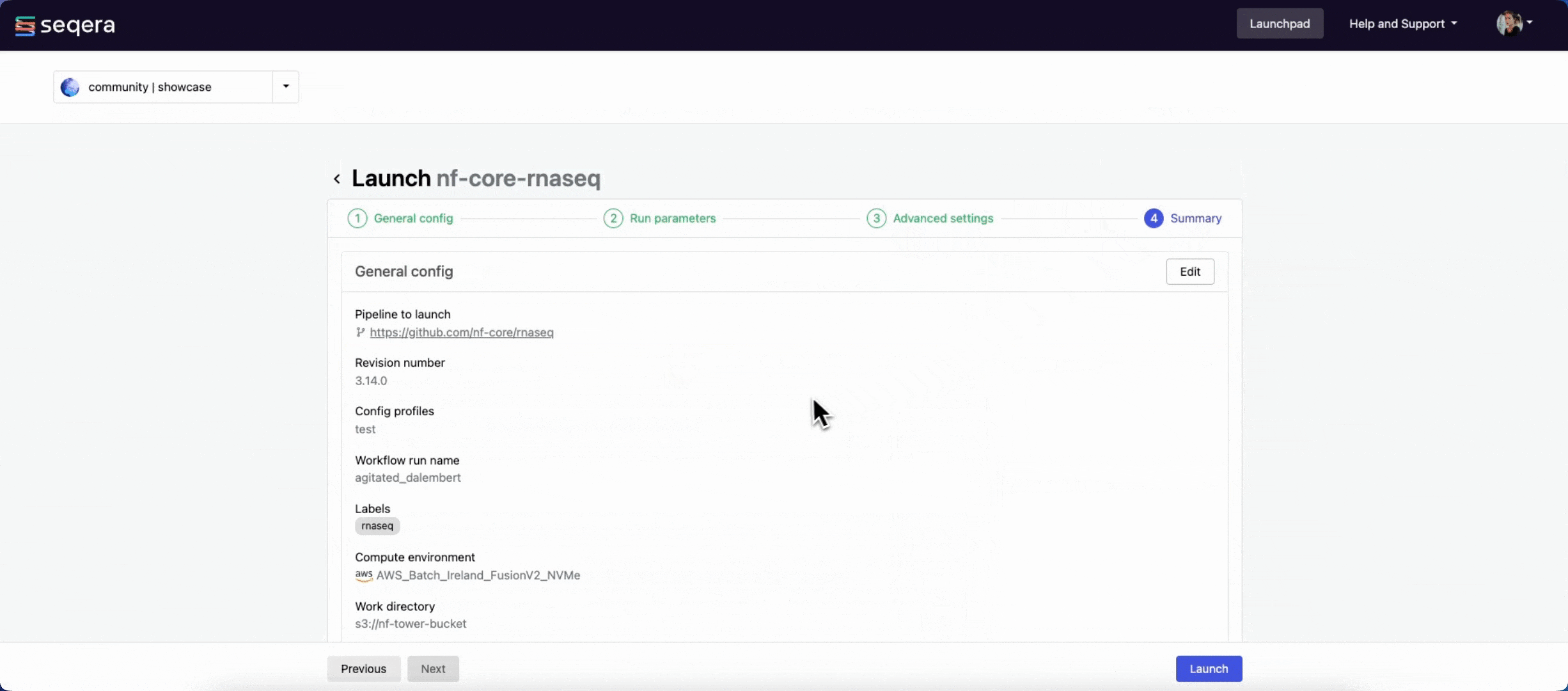
The launch form consists of General config, Run parameters, and Advanced options sections to specify your run parameters before execution, and an execution summary. Use section headings or select the Previous and Next buttons at the bottom of the page to move between sections.
Nextflow parameter schema
The launch form lets you configure the pipeline execution. The pipeline parameters in this form are rendered from a pipeline schema file in the root of the pipeline Git repository. nextflow_schema.json is a JSON-based schema that describes pipeline parameters. Pipeline developers use it to adapt their in-house Nextflow pipelines to run in Platform.
See Best Practices for Deploying Pipelines with the Seqera Platform to learn how to build the parameter schema for any Nextflow pipeline automatically with tooling maintained by the nf-core community.
General config
Most Showcase pipeline parameters are prefilled. Specify the following fields to identify your run among other workspace runs:
- Workflow run name: A unique identifier for the run, pre-filled with a random name. You can customize it.
- Labels: Assign new or existing labels to the run. For example, a project ID or genome version.
Run parameters
There are three ways to enter Run parameters prior to launch:
- The Input form view displays form fields to enter text, select attributes from drop-downs, and browse input and output locations with Data Explorer.
- The Config view displays a raw schema that you can edit directly. Select JSON or YAML format from the View as drop-down.
- Upload params file accepts a JSON or YAML file with run parameters.
input
Most nf-core pipelines use the input parameter in a standardized way to specify an input samplesheet that contains paths to input files (such as FASTQ files) and any additional metadata needed to run the pipeline. Use Browse to select either a file path in cloud storage via Data Explorer, or a pre-loaded Dataset:
- In the Data Explorer tab, select the
nf-tower-databucket, then search for and select thernaseq_sample_data.csvfile. - In the Datasets tab, search for and select
rnaseq_sample_data.
See Add data to learn how to add datasets and Data Explorer cloud buckets to your own workspaces.
output
Most nf-core pipelines use the outdir parameter to specify where the pipeline publishes final results. outdir must be unique for each pipeline run. Otherwise, your results are overwritten.
For this tutorial test run, keep the default outdir value (./results).
For the outdir parameter in pipeline runs in your own workspace, select Browse to specify a cloud storage directory using Data Explorer, or enter a cloud storage directory path to publish pipeline results to manually.
Pipeline-specific parameters
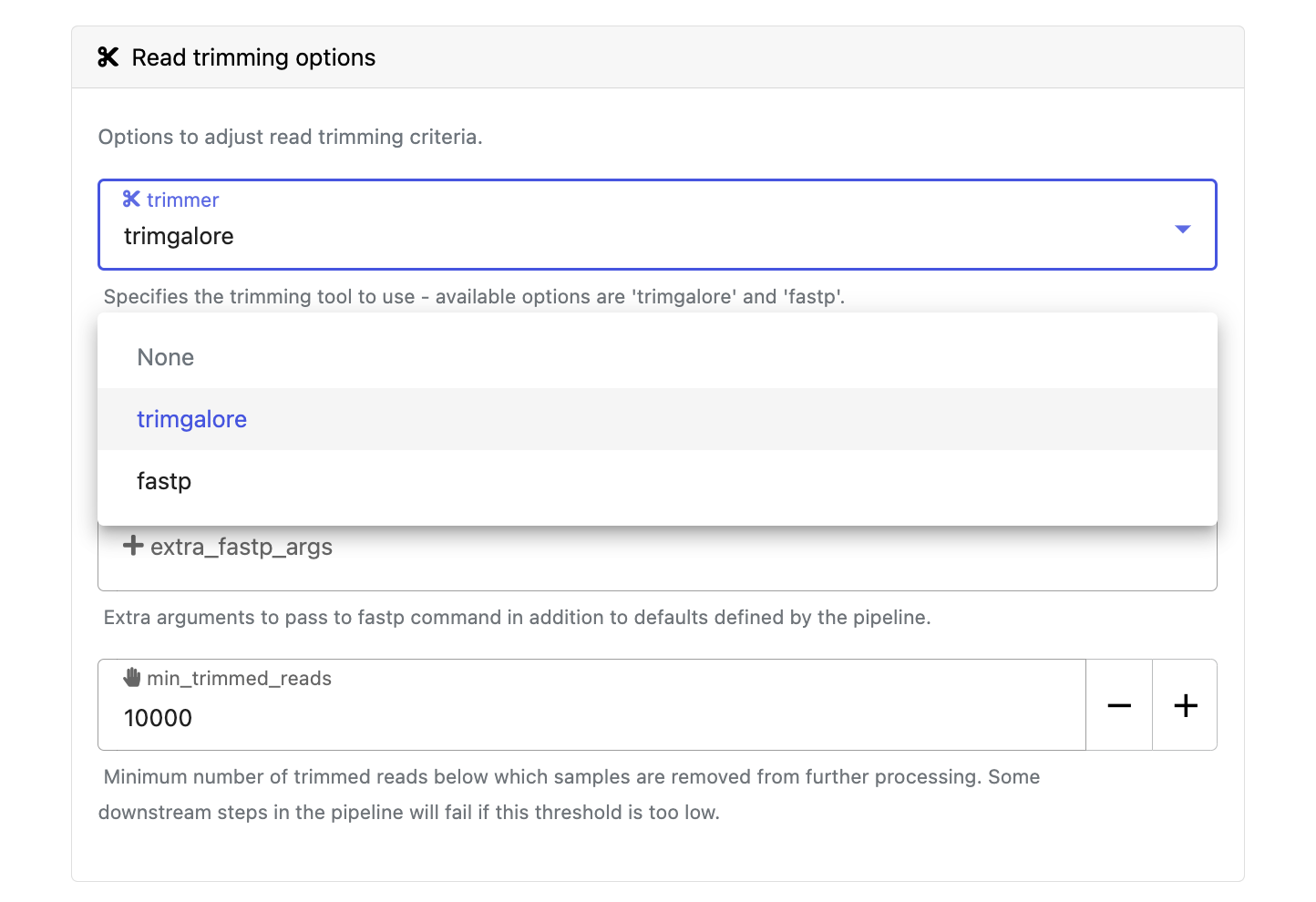
Modify other parameters to customize the pipeline execution through the parameters form. For example, under Read trimming options, change the trimmer to select fastp in the drop-down instead of trimgalore.
Select Launch to start the run and be directed to the Runs tab with your run in a submitted status at the top of the list.
View run information
Run details page
As the pipeline runs, the run details populate with parameters, logs, and other execution details:
View run details
- Command-line: The Nextflow command invocation used to run the pipeline. This contains details about the pipeline version (
-r 3.14.0flag) and profile, if specified (-profile testflag). - Parameters: The exact set of parameters used in the execution. This is helpful for reproducing the results of a previous run.
- Resolved Nextflow configuration: The full Nextflow configuration settings used for the run. This includes parameters, but also settings specific to task execution (such as memory, CPUs, and output directory).
- Execution Log: A summarized Nextflow log with information about the pipeline and the status of the run.
- Datasets: Link to datasets, if any were used in the run.
- Reports: View pipeline outputs directly in Platform.
View reports
Most Nextflow pipelines generate reports or output files worth inspecting at the end of a run. Reports can contain quality control (QC) metrics to assess the integrity of the results.
View run reports
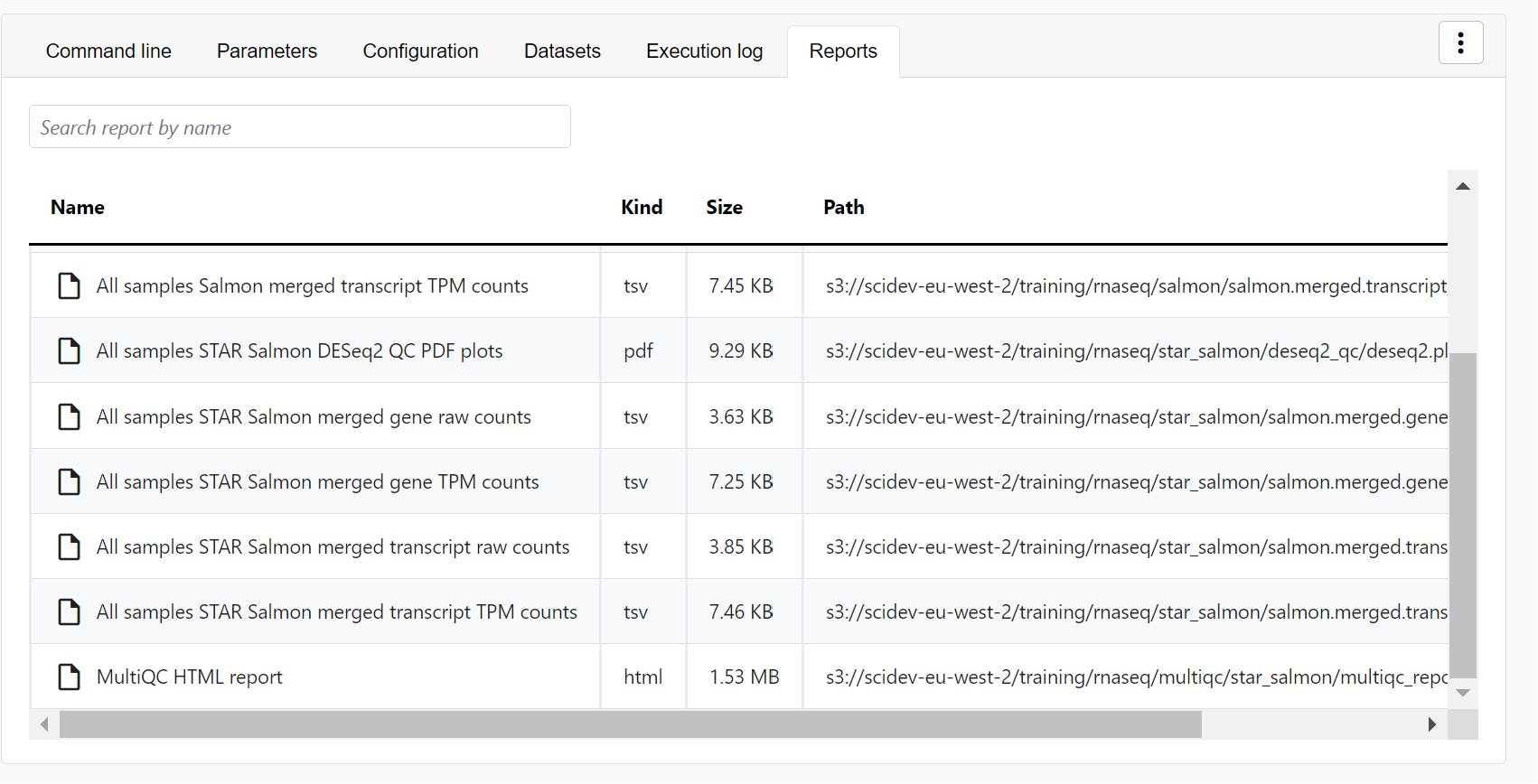
For example, for the nf-core/rnaseq pipeline, view the generated MultiQC report. MultiQC generates aggregate statistics and summaries from bioinformatics tools.
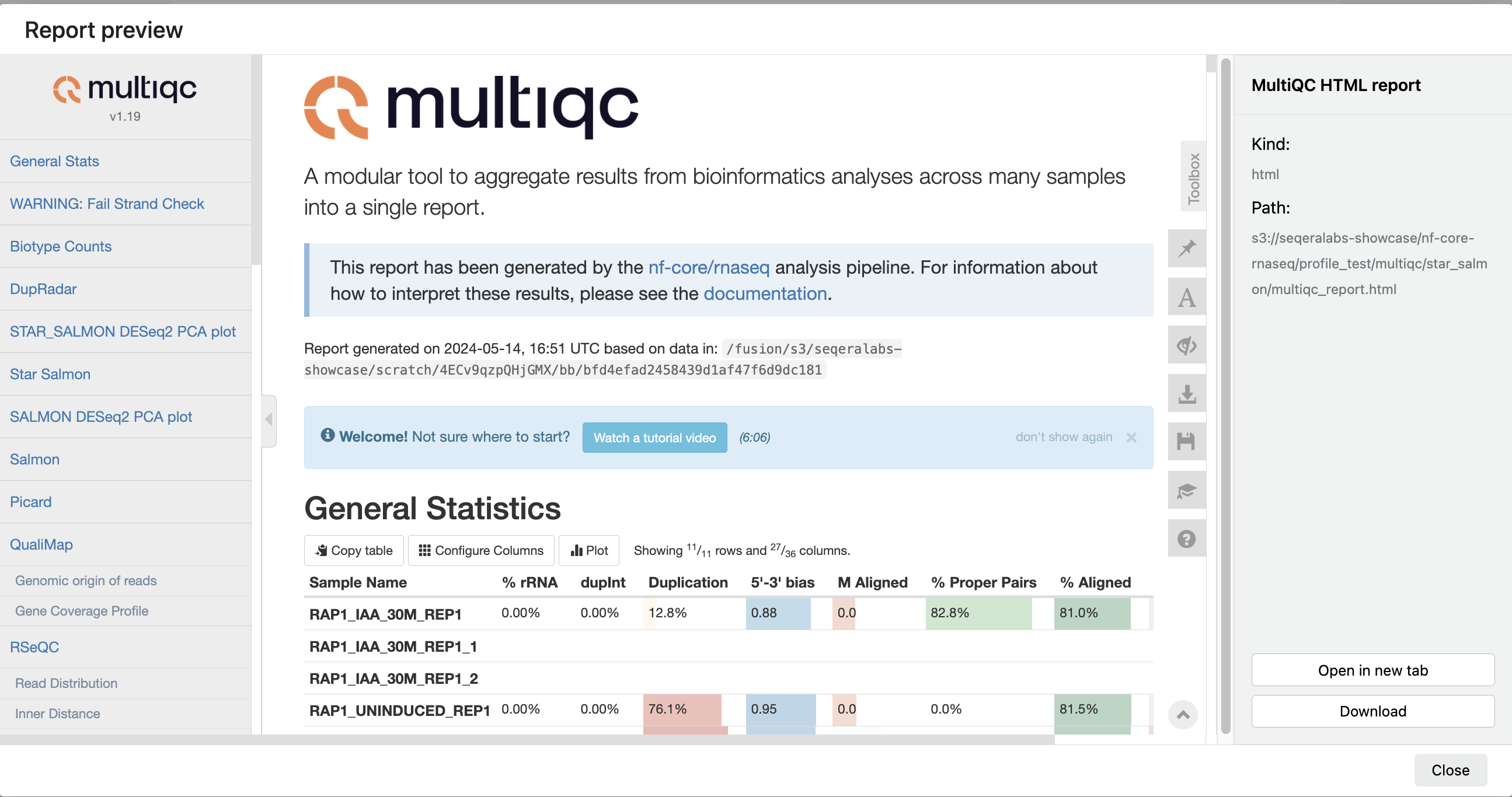
The paths to report files point to a location in cloud storage (in the outdir directory specified during launch), but you can view the contents directly and download each file without navigating to the cloud or a remote filesystem.
Specify outputs in reports
To tell Platform where to find the reports generated by the pipeline, include a tower.yml file that lists the report locations in the pipeline repository.
In the nf-core/rnaseq pipeline, the MULTIQC process step generates a MultiQC report file in HTML format:
reports:
multiqc_report.html:
display: "MultiQC HTML report"
See Reports to configure reports for pipeline runs in your own workspace.
View general information
The Run details page includes general information about who executed the run and when, the Git hash and tag used, and additional details about the compute environment and Nextflow version used.
View general run information
The General panel displays top-level information about a pipeline run:
- Unique workflow run ID
- Workflow run name
- Timestamp of pipeline start (timezones are based on system settings)
- Pipeline version and Git commit ID
- Nextflow session ID
- Username of the launcher
- Work directory path
View process and task details
Scroll down the page to view:
- The progress of individual pipeline Processes
- Aggregated stats for the run (total walltime, CPU hours)
- Workflow metrics (CPU efficiency, memory efficiency)
- A Task details table for every task in the workflow
The task details table provides further information on every step in the pipeline, including task statuses and metrics:
View task details
Select a task in the task table to open the Task details dialog. The dialog has three tabs: About, Execution log, and Data Explorer.
About
The About tab includes:
- Name: Process name and tag
- Command: Task script, defined in the pipeline process
- Status: Exit code, task status, and number of attempts
- Work directory: Directory where the task was executed
- Environment: Environment variables that were supplied to the task
- Execution time: Metrics for task submission, start, and completion time (timezones are based on system settings)
- Resources requested: Metrics for the resources requested by the task
- Resources used: Metrics for the resources used by the task
Execution log
The Execution log tab provides a real-time log of the selected task's execution. You can download task execution and other logs (such as stdout and stderr) here, if they remain in your compute environment.
Task work directory in Data Explorer
If a task fails, a good place to begin troubleshooting is the task's work directory. Nextflow hash-addresses each task of the pipeline and creates unique directories based on these hashes.
View task log and output files
Instead of navigating through a bucket on the cloud console or filesystem, use the Data Explorer tab in the Task window to view the work directory.
Data Explorer shows the log files and output files generated for each task, directly within Platform. You can view, download, and copy the link for these intermediate files to simplify troubleshooting.
Interactive analysis
Interactive analysis of pipeline results often happens in platforms like Jupyter Notebooks or the R-IDE. Setting up the infrastructure for these platforms, including access to pipeline data and the necessary bioinformatics packages, can be complex and time-consuming.
Studios simplifies creating interactive analysis environments. With built-in templates, creating a Studio is much like adding and sharing pipelines or datasets.
Analyze RNAseq data in Studios
In the Studios tab, you can monitor the Studios in the Community Showcase workspace and view their details.
Use Studios to perform custom analysis on the results of upstream pipelines. For example, in the Community Showcase workspace we ran the nf-core/rnaseq pipeline to quantify gene expression, followed by nf-core/differentialabundance to derive differential expression statistics. The workspace contains a Studio with these results mounted from cloud storage for further analysis. One of these outputs is an RShiny application, which you can deploy for interactive analysis.
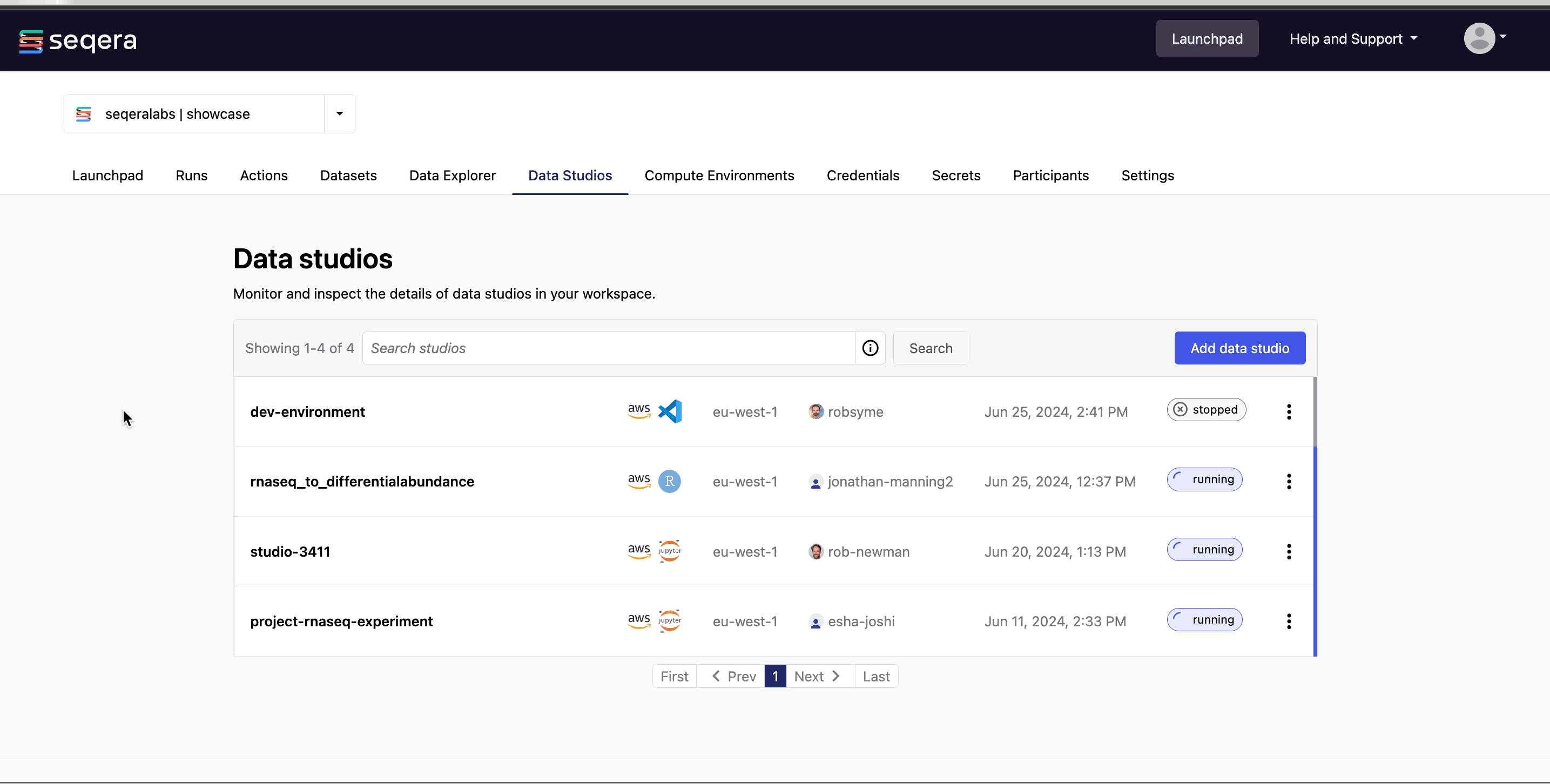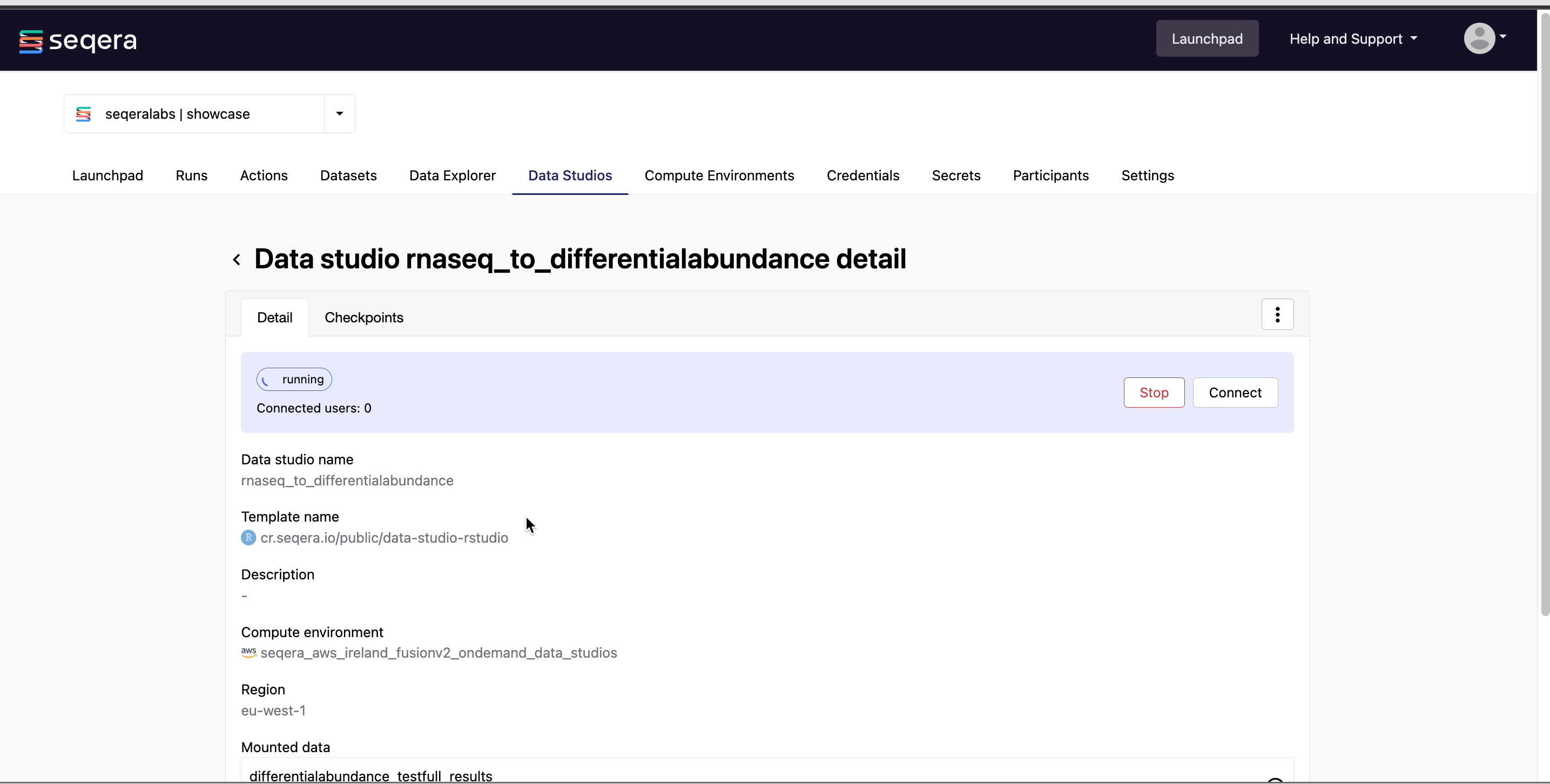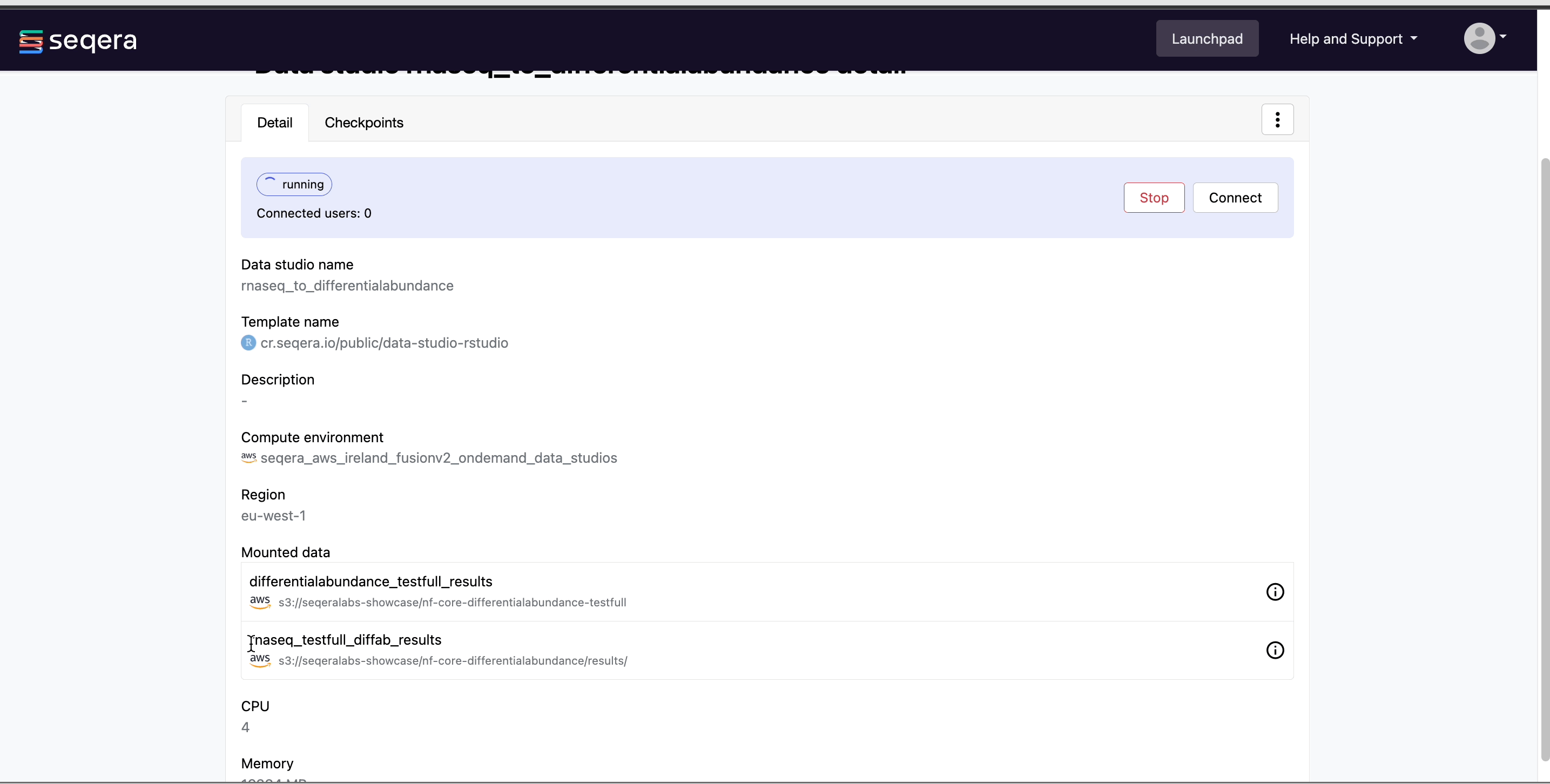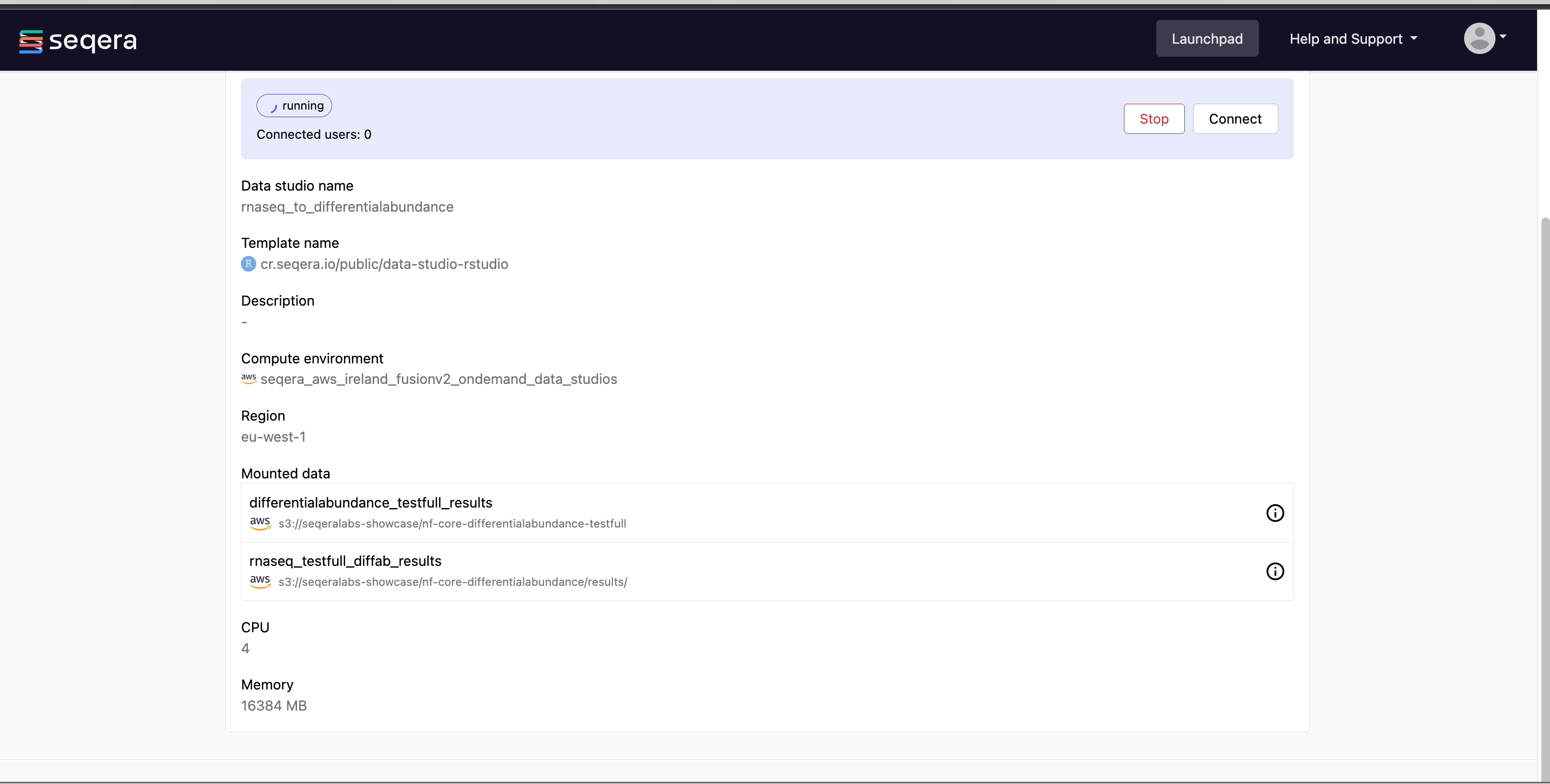
Connect to the RNAseq analysis Studio
Select the rnaseq_to_differentialabundance Studio. This Studio consists of an R-IDE that uses an existing compute environment in the Community Showcase workspace. The Studio also contains mounted data generated from the nf-core/rnaseq and subsequent nf-core/differentialabundance pipeline runs, directly from AWS S3.
Select Connect to view the running R-IDE session. The rnaseq_to_differentialabundance Studio includes the necessary R packages for deploying a web app to visualize the RNAseq data.
Deploy the RShiny app in the Studio by selecting the play button on the last chunk of the R script:
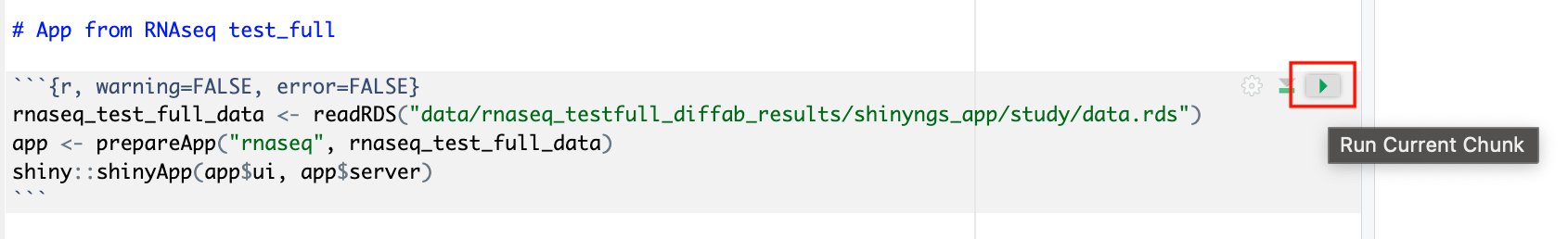
You can specify the resources each Studio uses. When you create your own Studios with shared compute environment resources, you must allocate sufficient resources to the compute environment to prevent Studio or pipeline run interruptions.
Explore results
The RShiny app deploys in a separate browser window with a data interface. Here you can view information about your sample data, perform QC or exploratory analysis, and view the results of differential expression analyses.
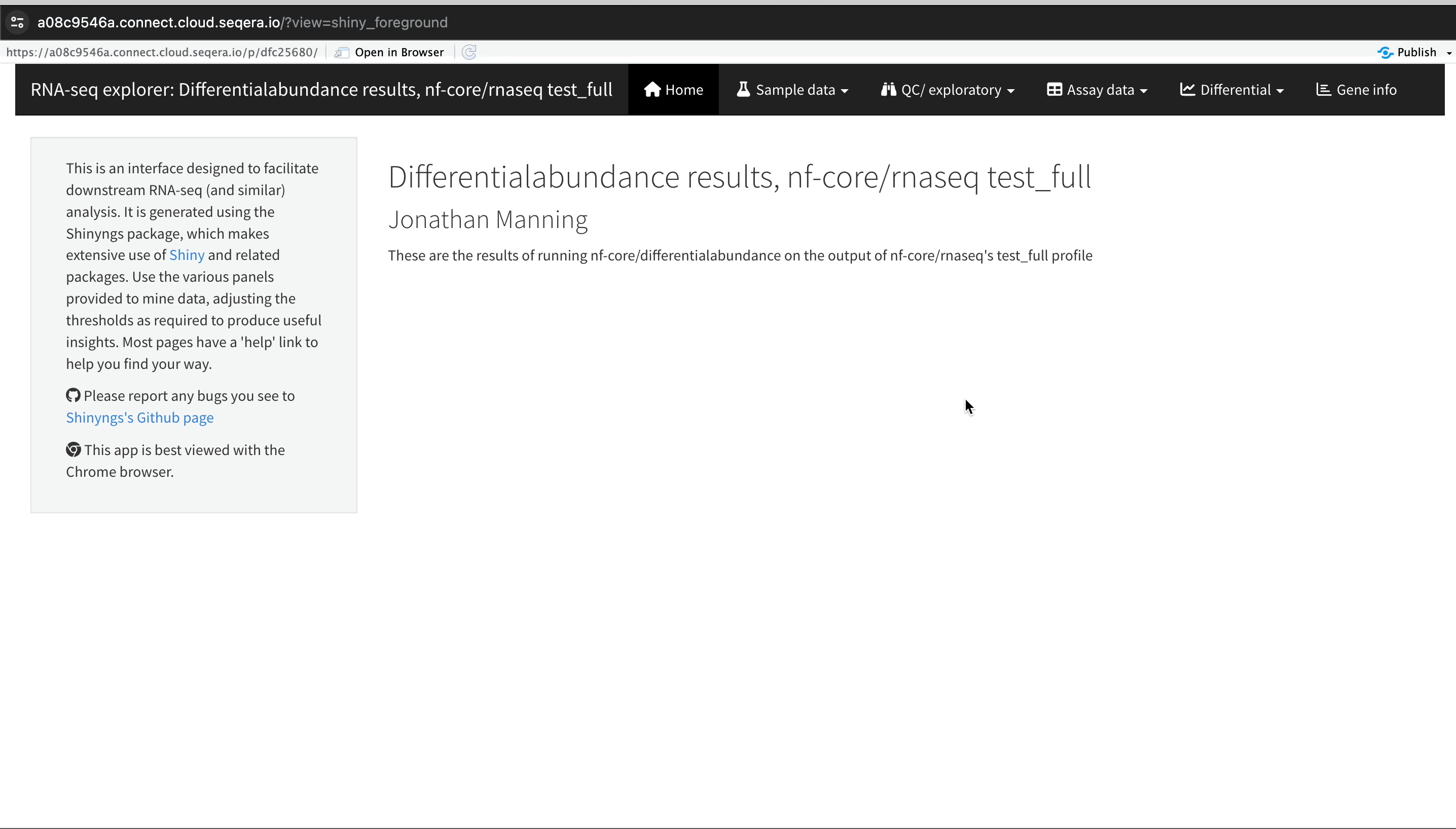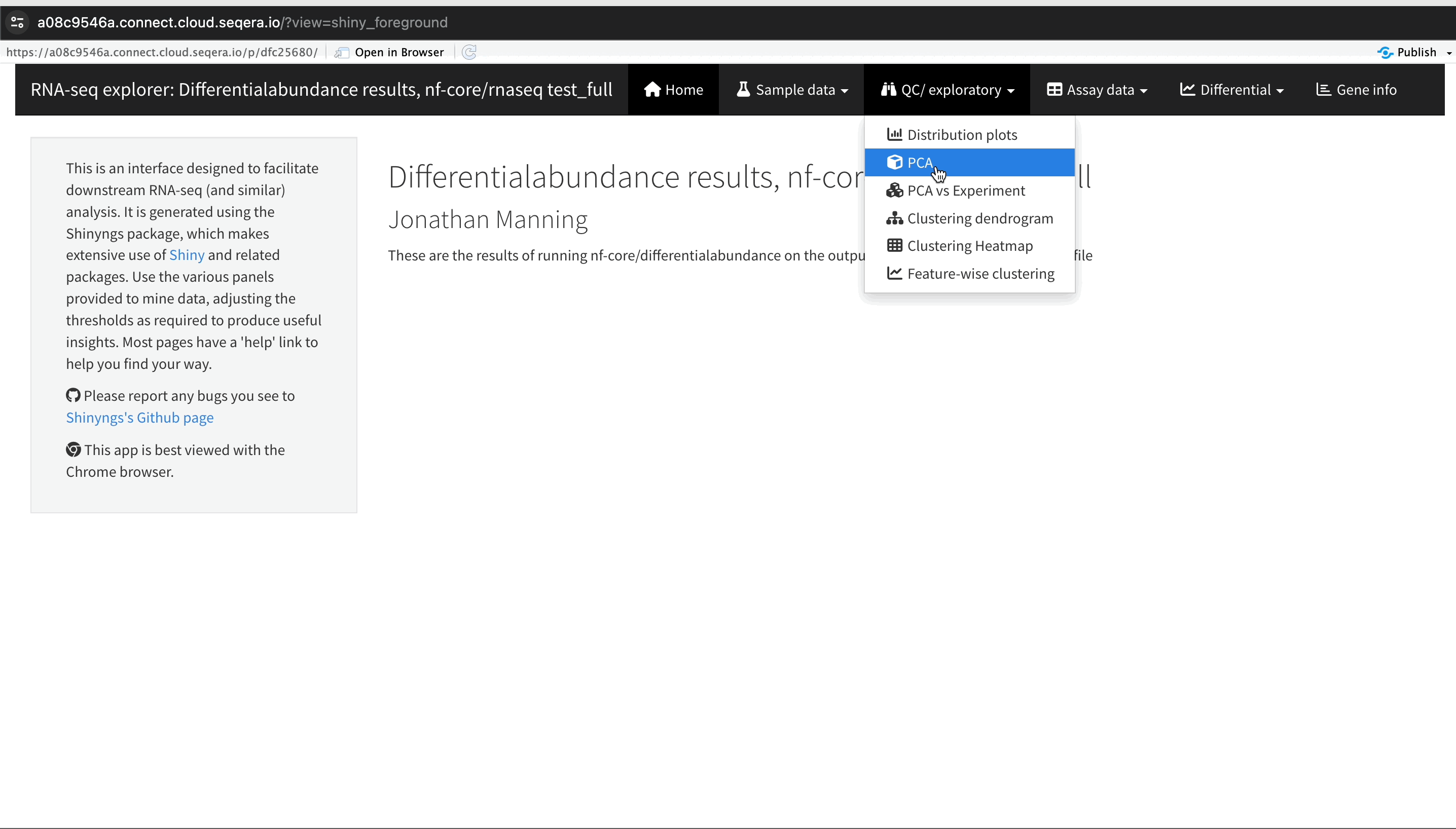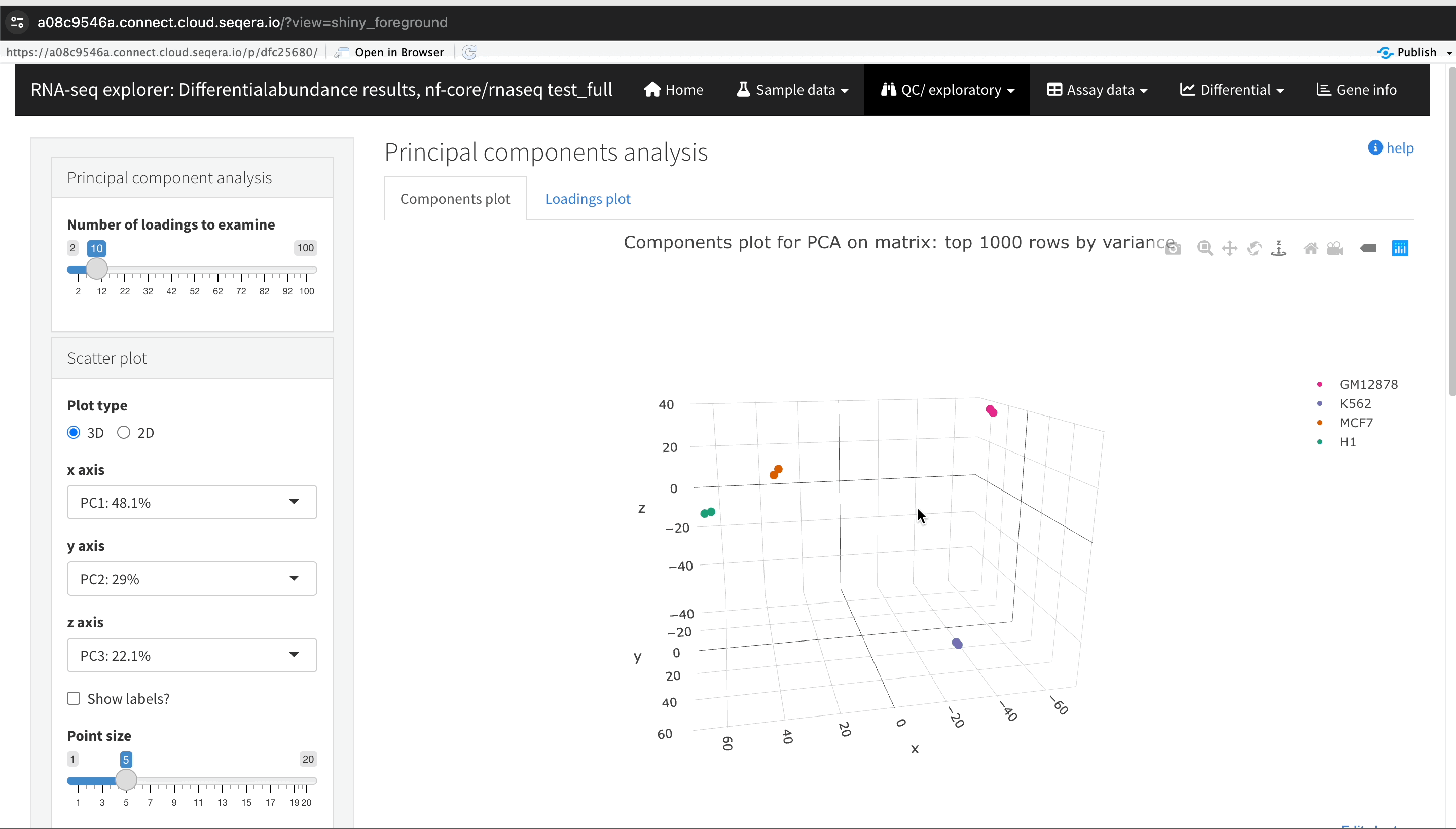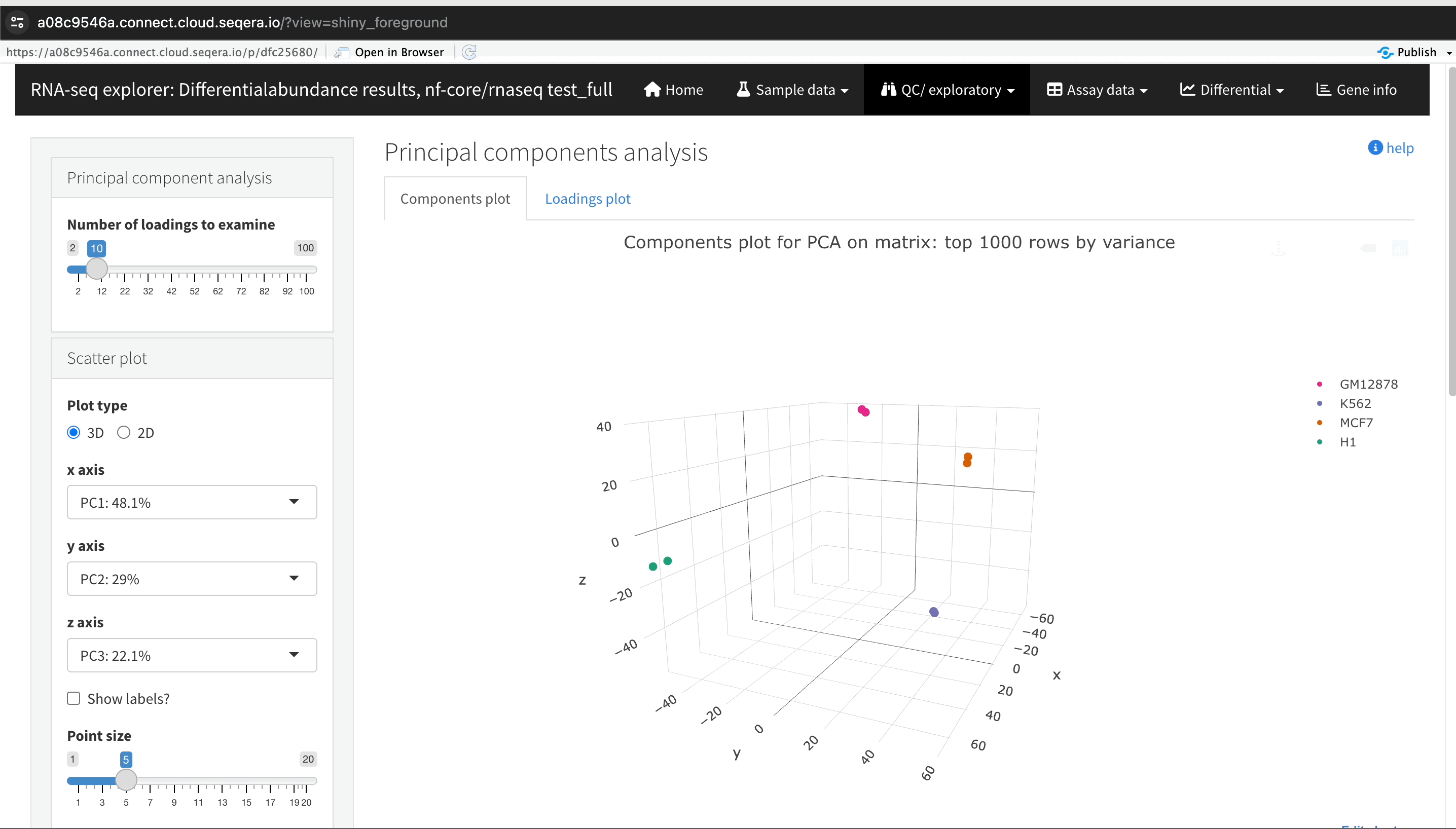
Sample clustering with PCA plots
In the QC/Exploratory tab, select the PCA (Principal Component Analysis) plot to visualize how the samples group together based on their gene expression profiles.
In this example, we used RNA sequencing data from the publicly available ENCODE project, which includes samples from four different cell lines:
- GM12878: a lymphoblastoid cell line
- K562: a chronic myelogenous leukemia cell line
- MCF-7: a breast cancer cell line
- H1-hESC: human embryonic stem cells
What to look for in the PCA plot:
- Replicate clustering: Ideally, biological replicates of the same cell type should cluster closely together. For example, replicates of MCF-7 (breast cancer cell line) group together. This indicates consistent gene expression profiles among biological replicates.
- Cell type separation: Different cell types should form distinct clusters. For instance, GM12878, K562, MCF-7, and H1-hESC samples should each form their own separate clusters, reflecting their unique gene expression patterns.
From this PCA plot, you can assess the consistency and quality of your sequencing data, identify potential issues, and understand the major sources of variation among your samples, all directly in Platform.
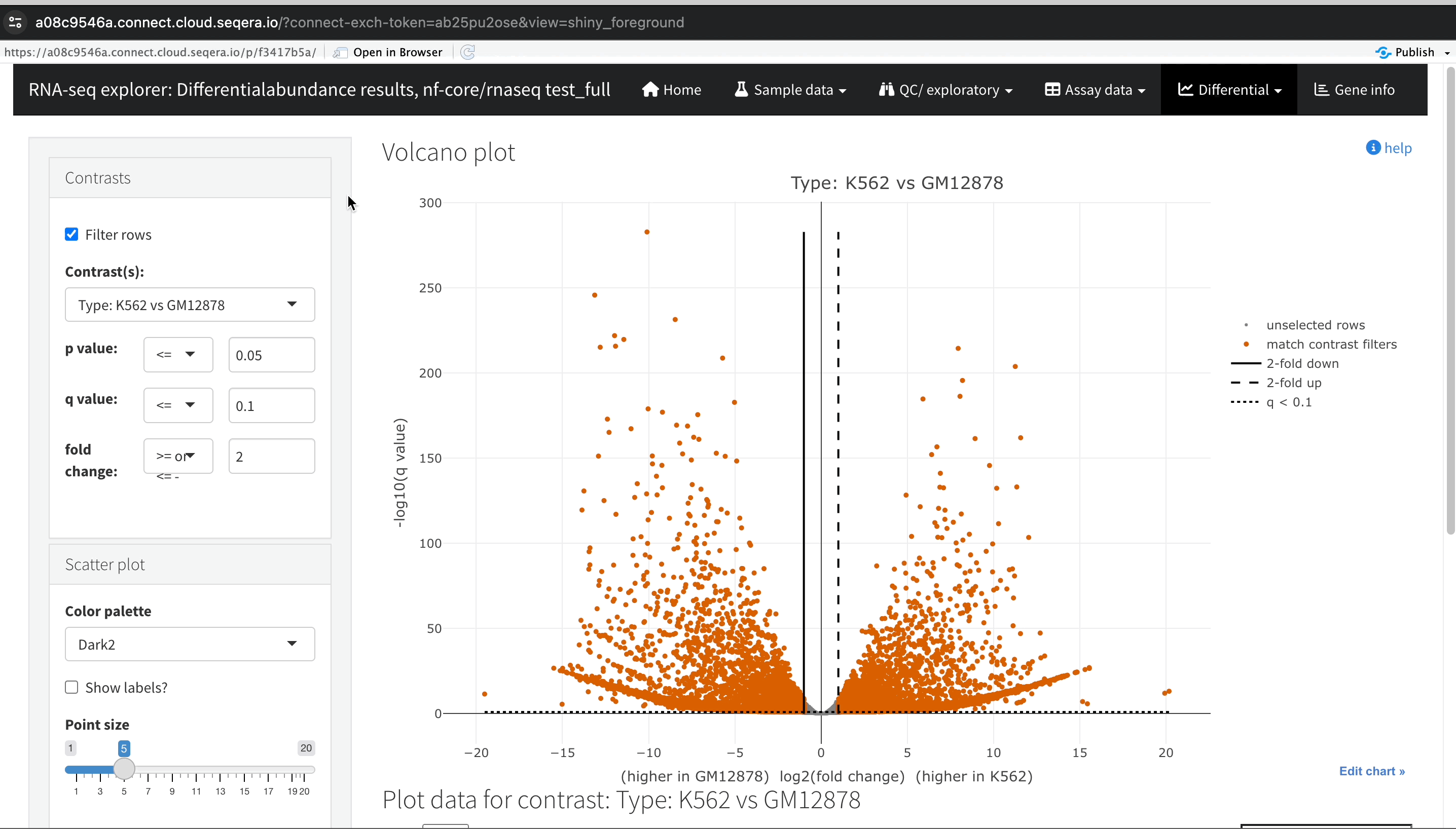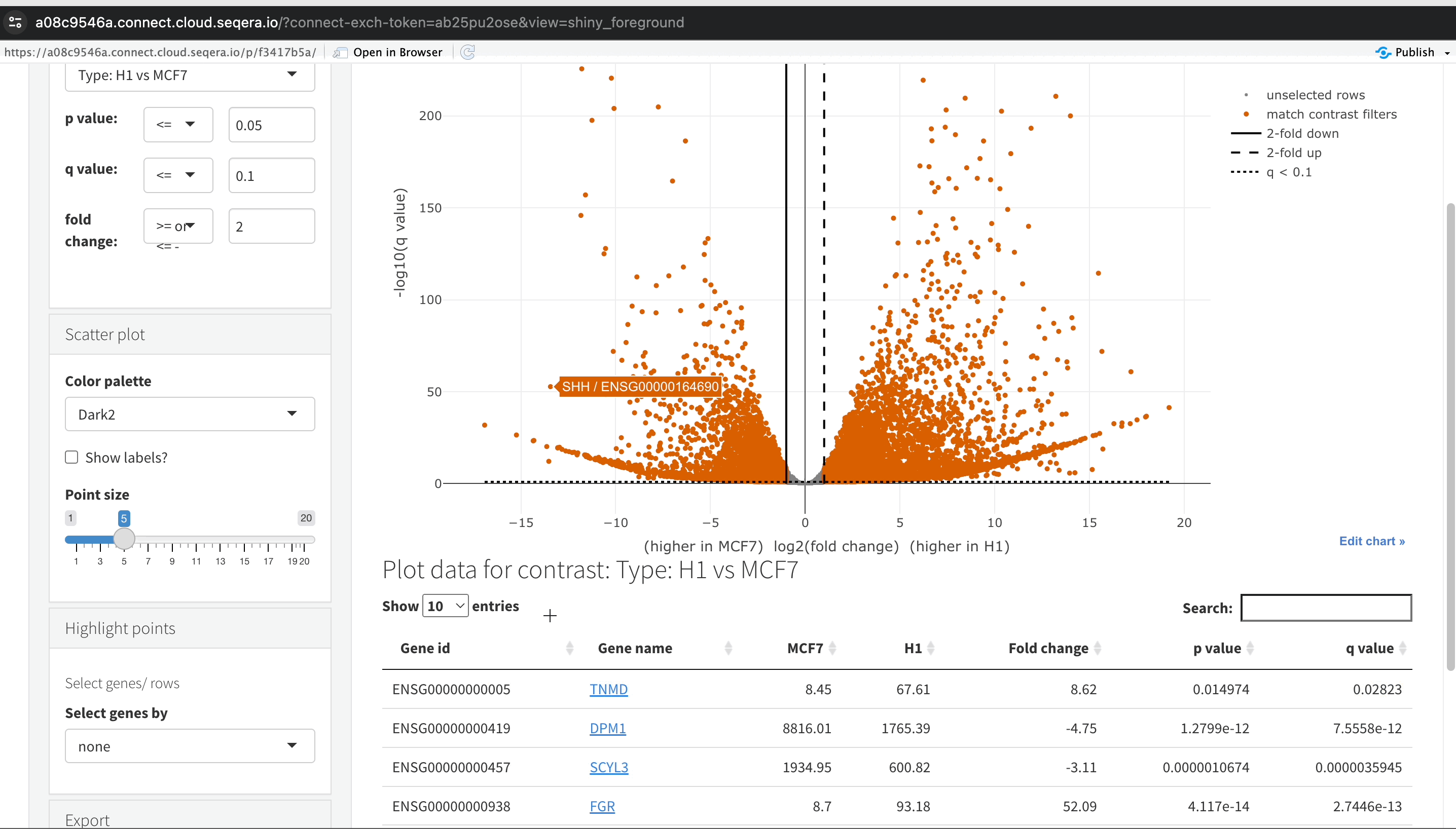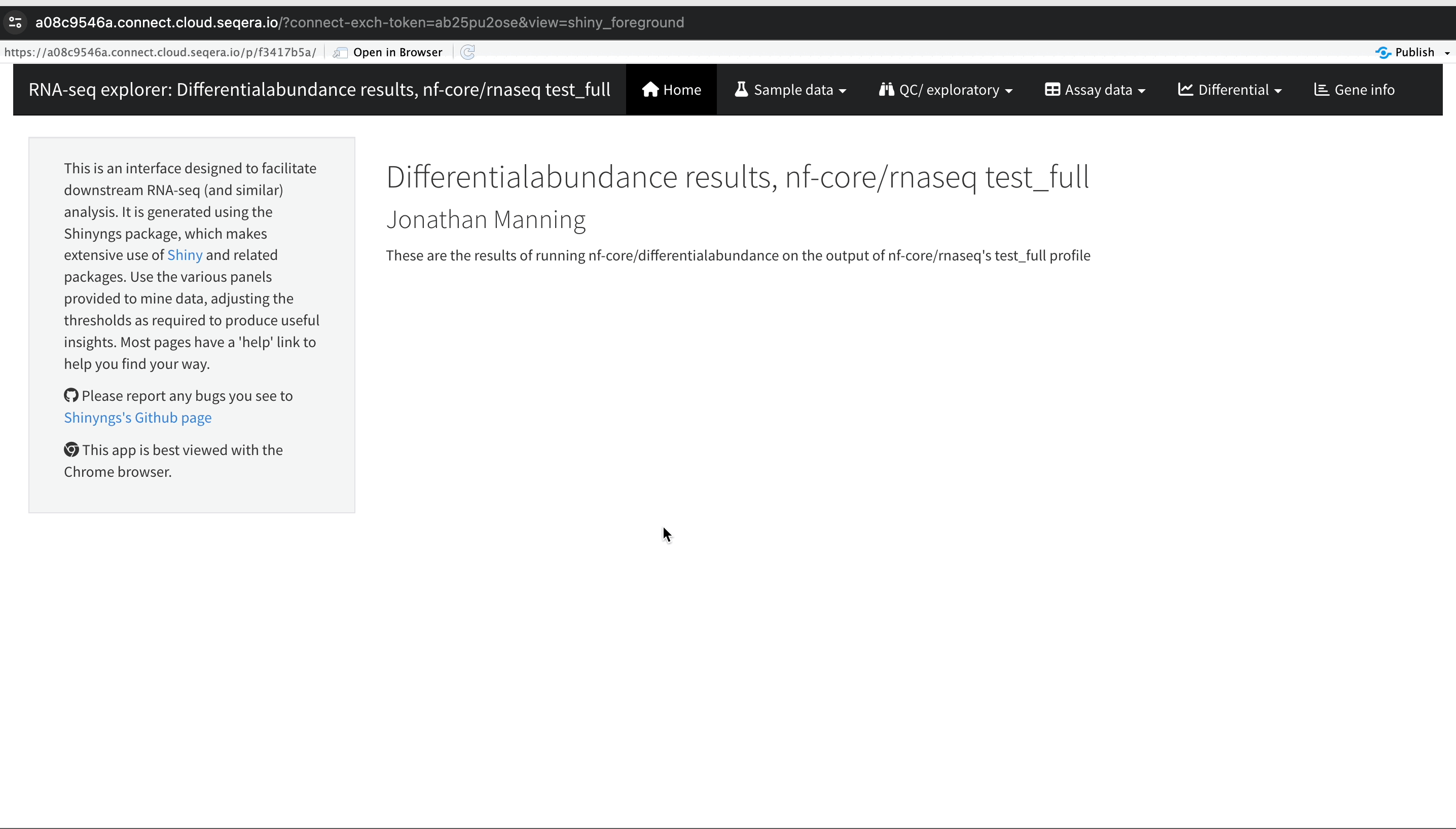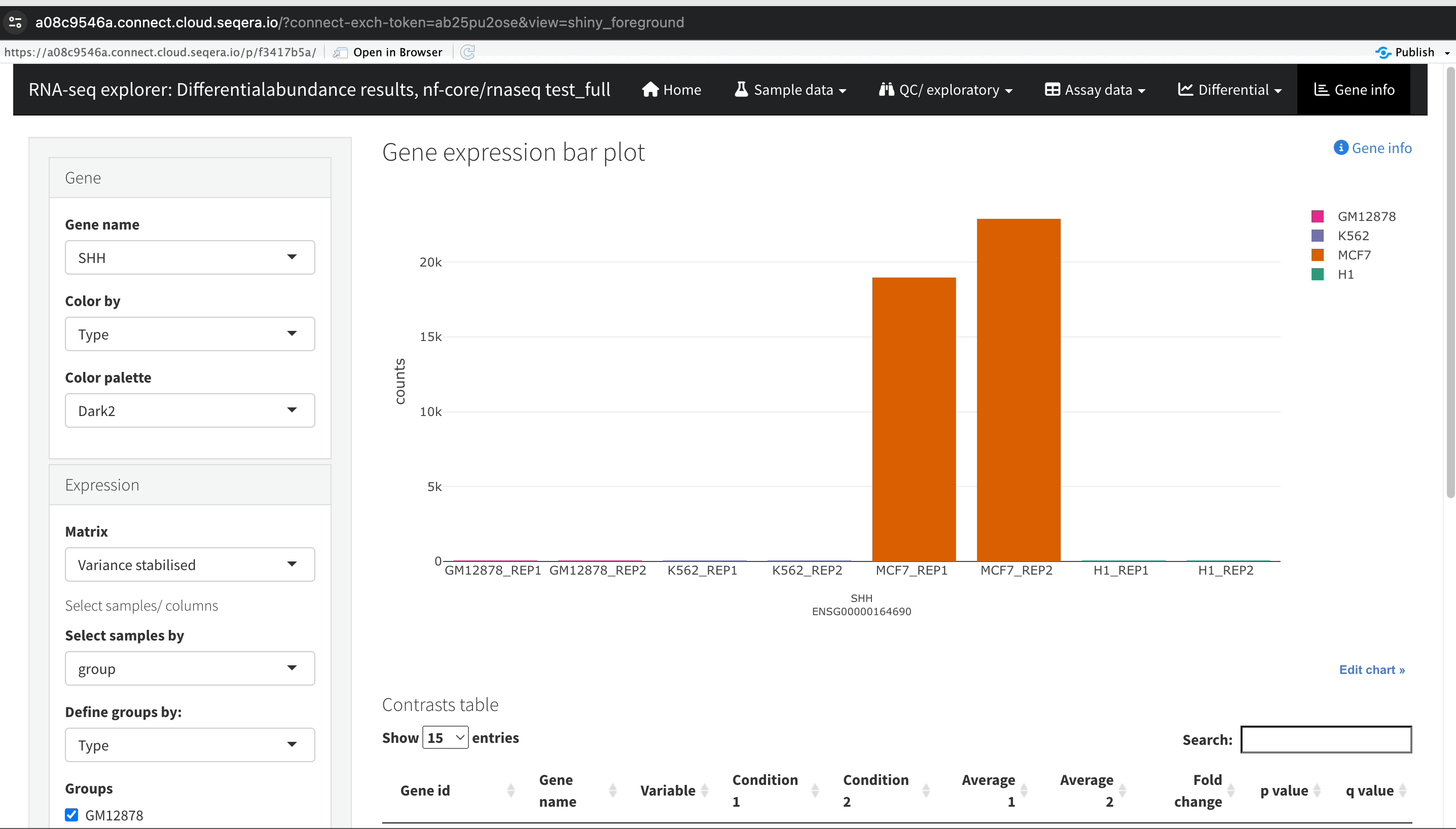
Gene expression changes with Volcano plots
In the Differential tab, select Volcano plots to compare genes with significant changes in expression between two samples. For example, filter for Type: H1 vs MCF-7 to view the differences in expression between these two cell lines.
-
Identify upregulated and downregulated genes: The x-axis of the volcano plot represents the log2 fold change in gene expression between the H1 and MCF-7 samples, while the y-axis represents the statistical significance of the changes.
- Upregulated genes in MCF-7: Genes on the left side of the plot (negative fold change) are upregulated in the MCF-7 samples compared to H1. For example, the SHH gene, which is known to be upregulated in cancer cell lines, prominently appears here.
-
Filtering for specific genes: To focus on specific genes, use the filter. For example, filter for the SHH gene in the table below the plot to locate and examine it in more detail.
-
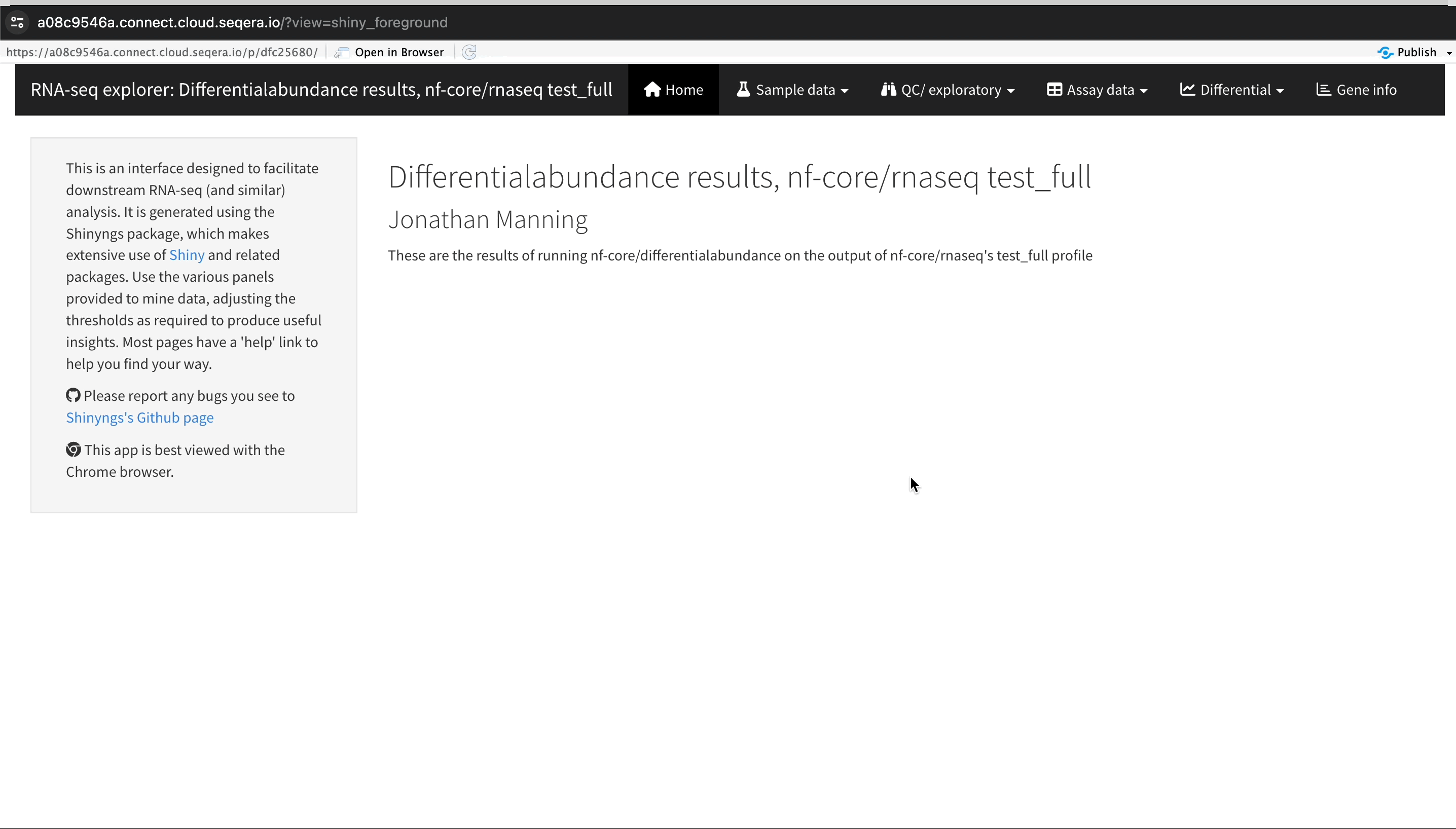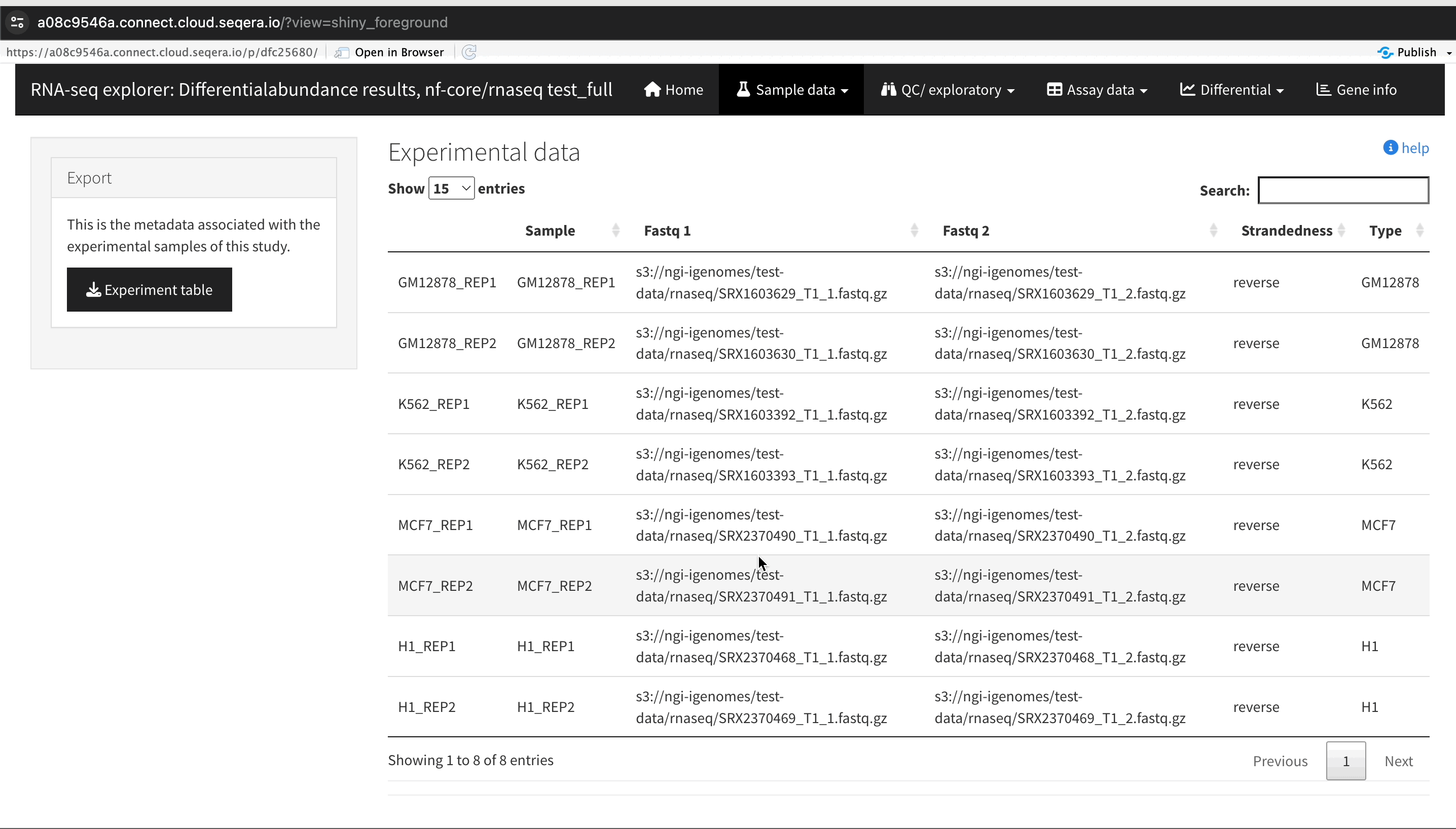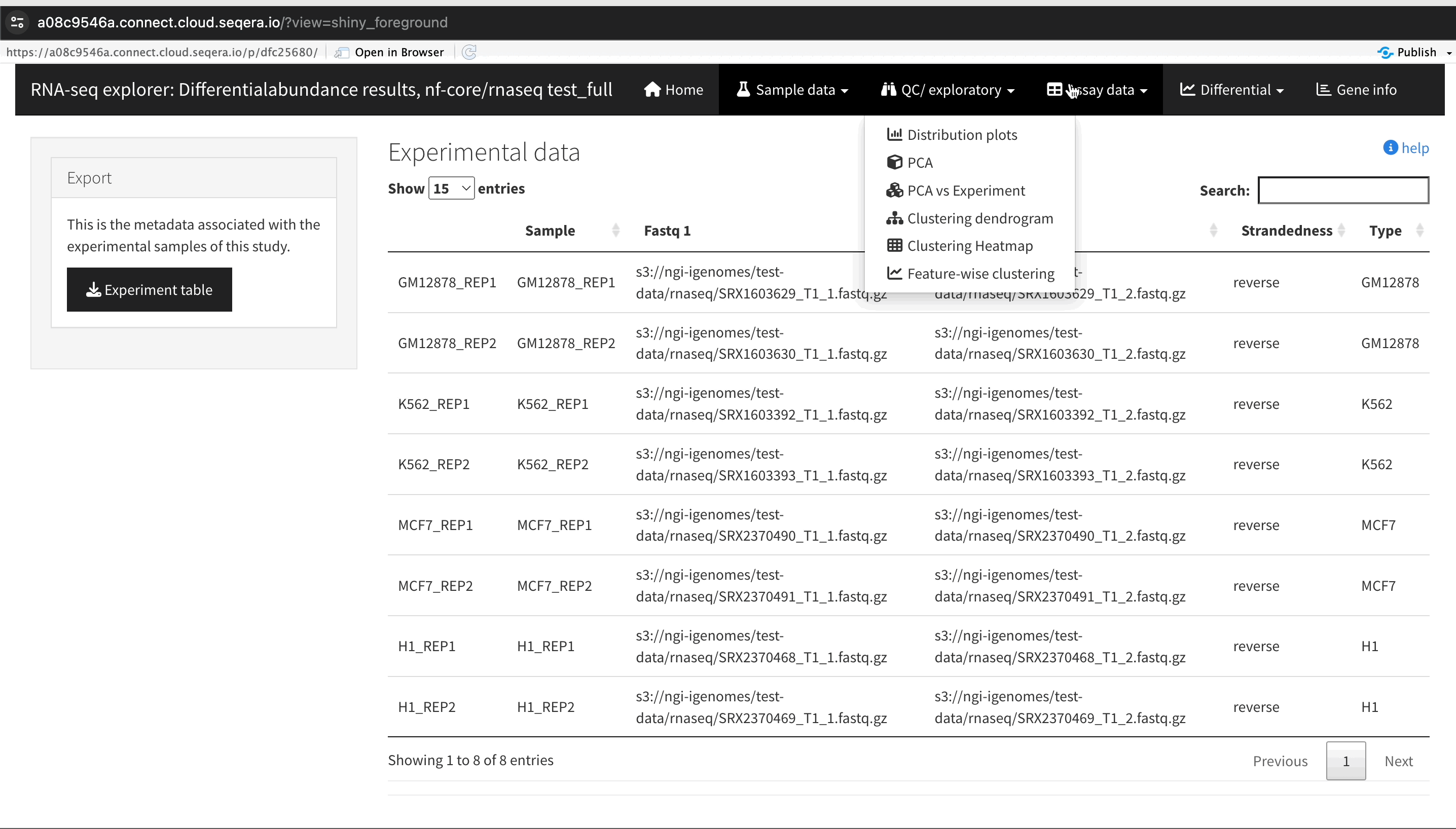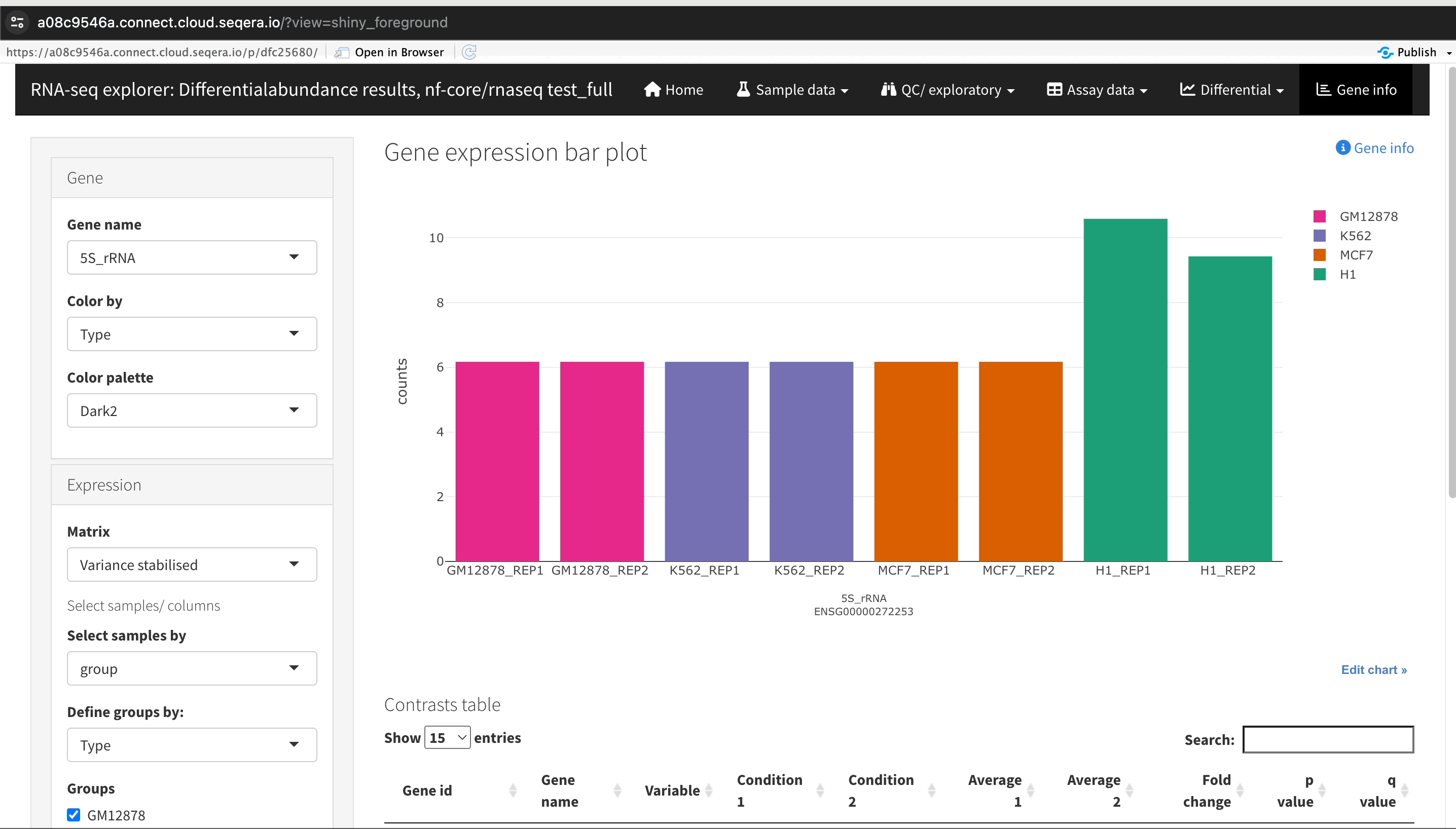
Gene expression bar plot: After filtering for the SHH gene, select it to open a gene expression bar plot. This plot shows the expression levels of SHH across all samples and where it is most highly expressed.
- Here, SHH is most highly expressed in MCF-7, which aligns with its known role in cancer cell proliferation.
The volcano plot helps you identify and explore the genes with the largest expression changes between your samples.
Collaborate in the Studio
To share your RNAseq analysis results or let colleagues run their own exploratory analysis, select the options menu for the Studio, then select Copy Studio URL. With this link, other authenticated users with the Connect role (or greater) can access the session directly.
See Studios to learn how to create Studios in your own workspace.
Pipeline optimization
The task-level resource usage metrics in Seqera Platform show the resources requested for a task and what it actually used. This information helps you fine-tune your configuration.
However, manually adjusting resources for every task in your pipeline is impractical. Instead, use the pipeline optimization feature on the Launchpad.
Pipeline optimization analyzes resource usage data from previous runs to optimize the resource allocation for future runs. After a successful run, optimization becomes available, indicated by the lightbulb icon next to the pipeline turning black.
Optimize nf-core/rnaseq
Return to the Launchpad and select the lightbulb icon next to the nf-core/rnaseq pipeline to view the optimized profile. You can tailor the optimization's target settings and add a retry strategy as needed.
View optimized configuration
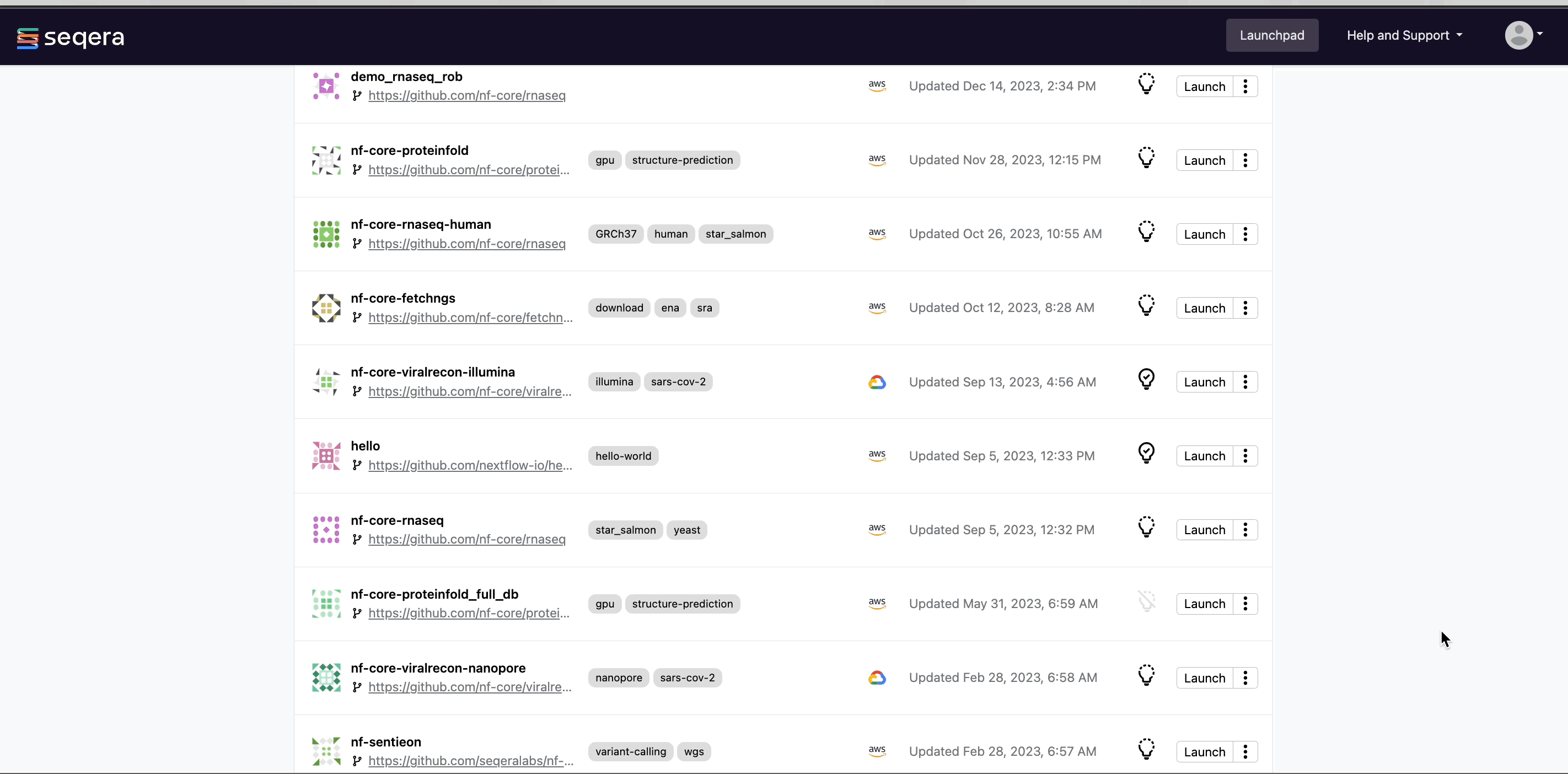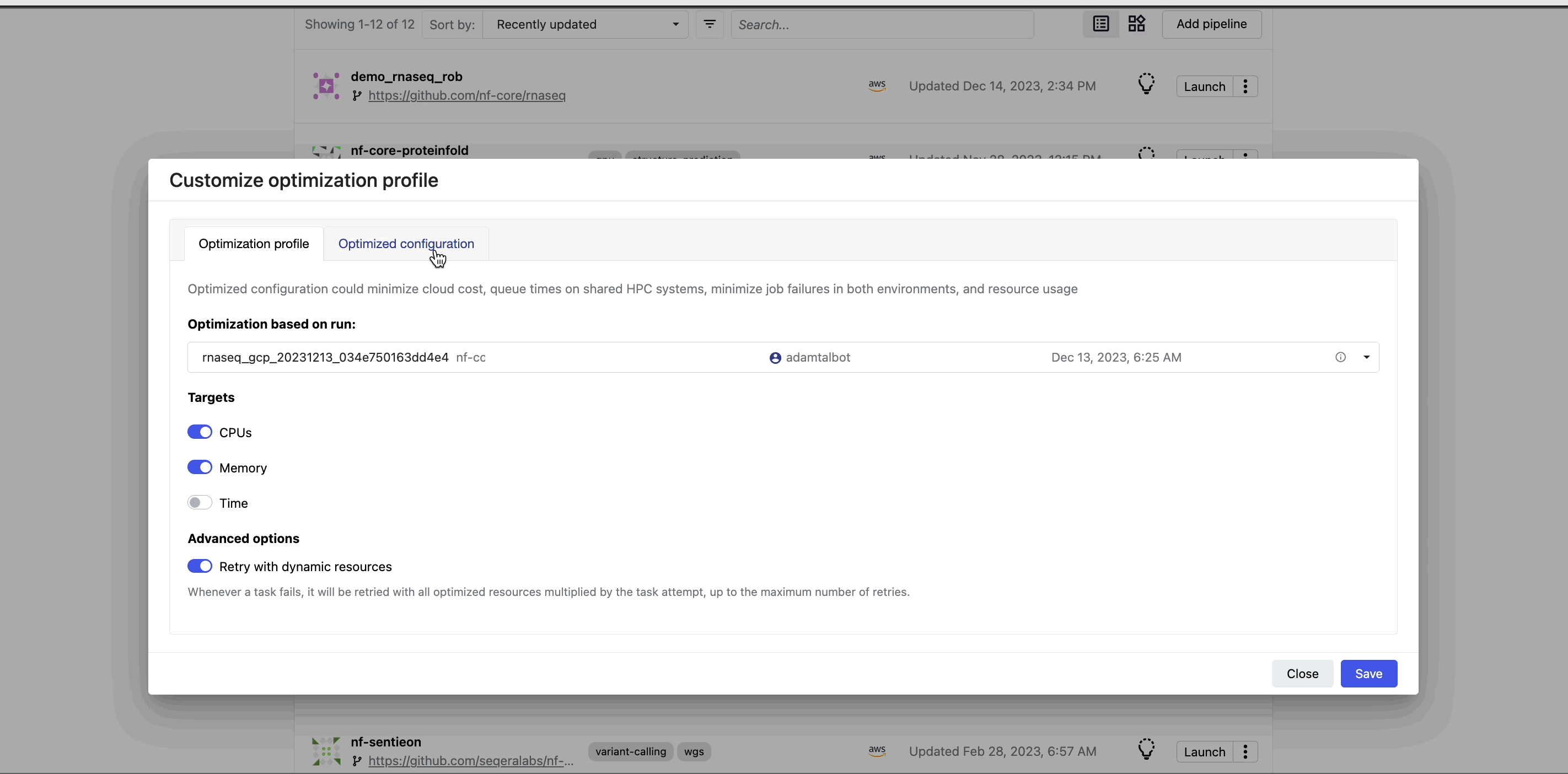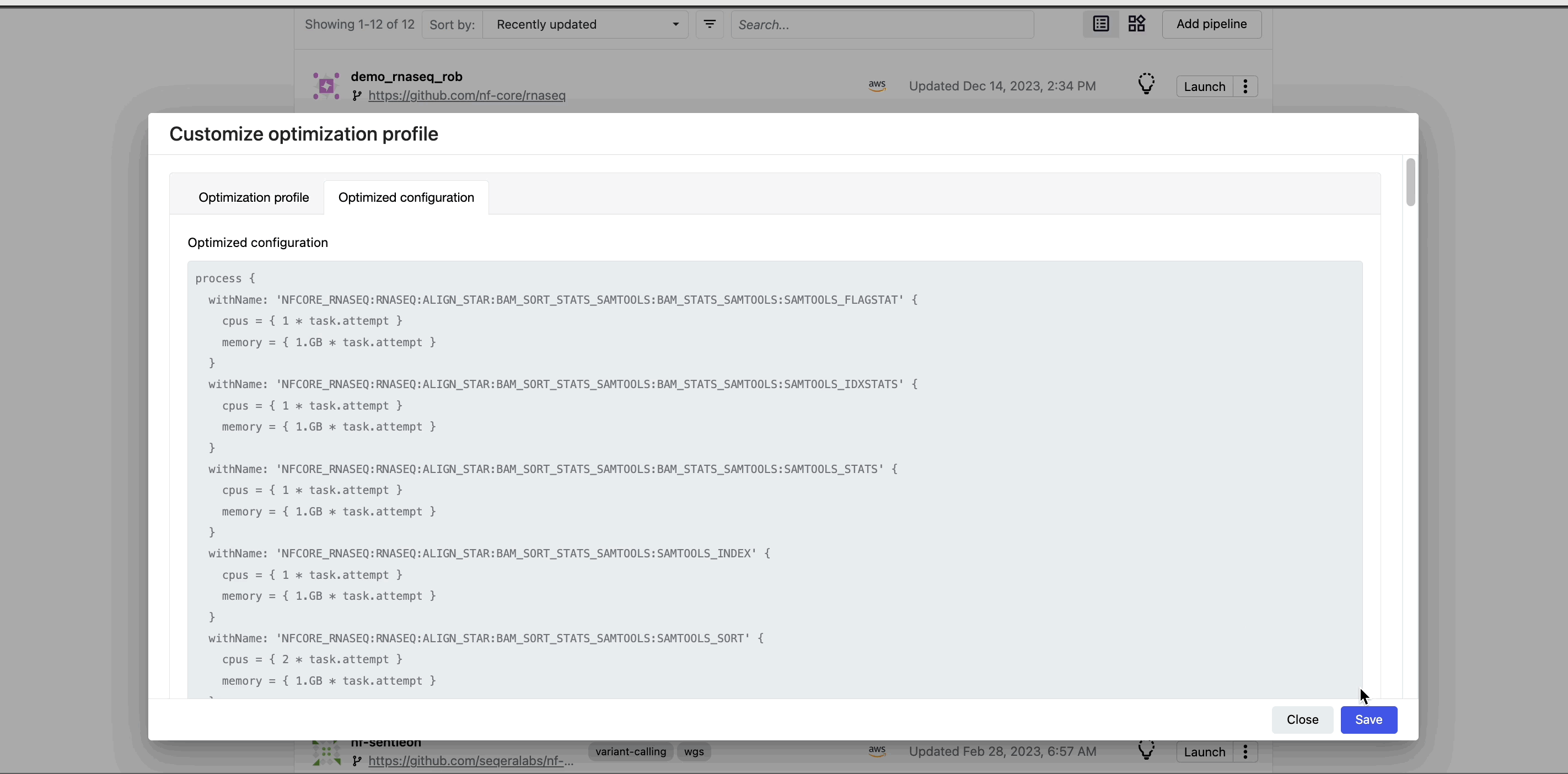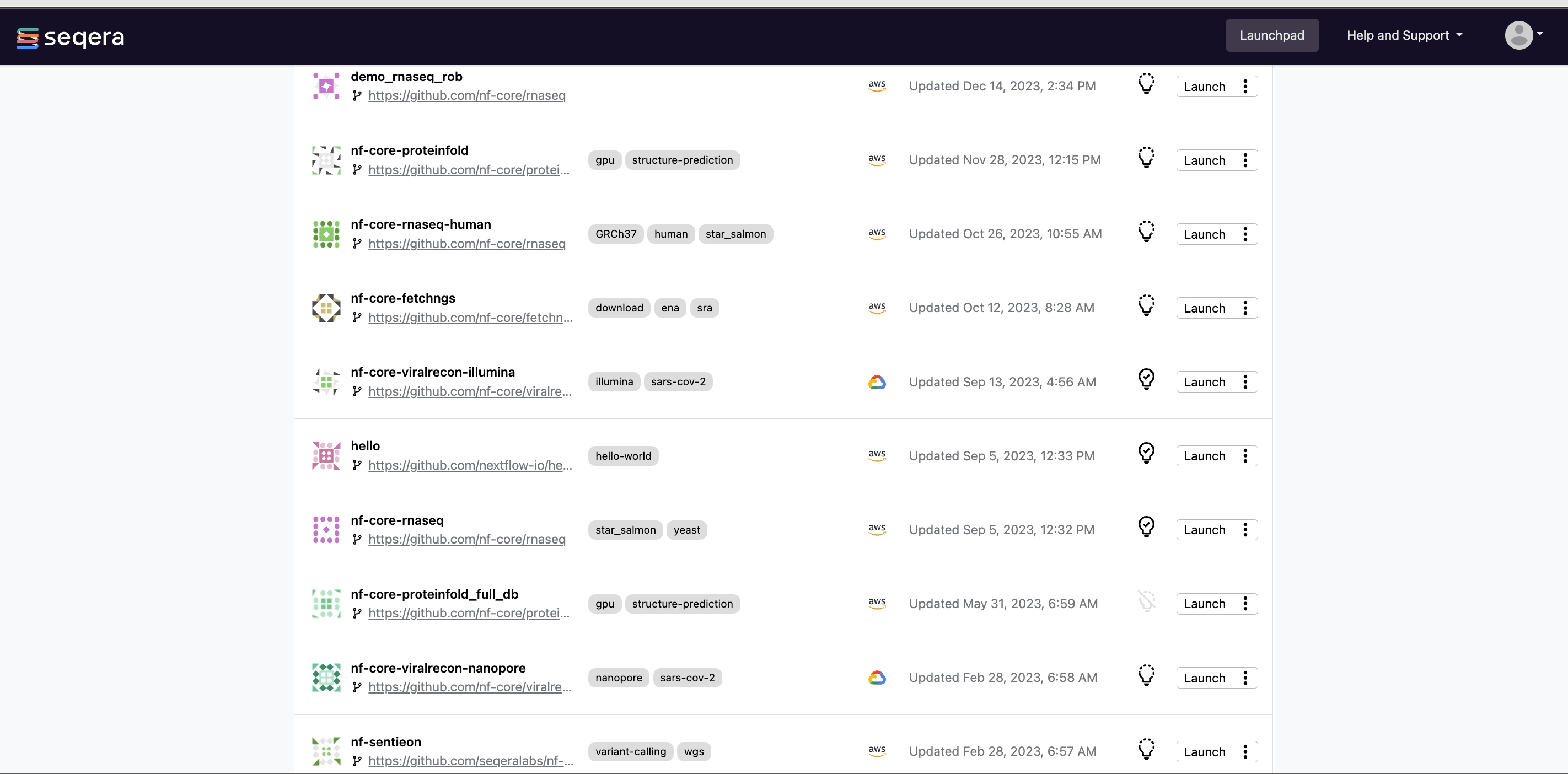
When you select the lightbulb, you can access an optimized configuration profile in the second tab of the Customize optimization profile window.
This profile consists of Nextflow configuration settings for each process and each resource directive (where applicable): cpus, memory, and time. The optimized setting for a given process and resource directive is based on the maximum use of that resource across all tasks in that process.
Once you select optimization, subsequent runs of that pipeline inherit the optimized configuration profile, indicated by the black lightbulb icon with a checkmark.
Optimization profiles are generated from one run at a time, defaulting to the most recent run, and not an aggregation of previous runs.
Verify the optimized configuration of a given run by inspecting the resource usage plots for that run and these fields in the run's task table:
| Description | Key |
|---|---|
| CPU usage | pcpu |
| Memory usage | peakRss |
| Runtime | start and complete |