Add pipelines
The Launchpad lists the preconfigured Nextflow pipelines that can be executed on the compute environments in your workspace.
Platform offers two methods to import pipelines to your workspace Launchpad — directly from Seqera Pipelines or manually via Add pipeline in Platform.
Import from Seqera Pipelines
Seqera Pipelines is a curated collection of quality open-source pipelines that can be imported directly to your workspace Launchpad in Platform. Each pipeline includes a dataset to use in a test run to confirm compute environment compatibility in just a few steps.
To import a pipeline:
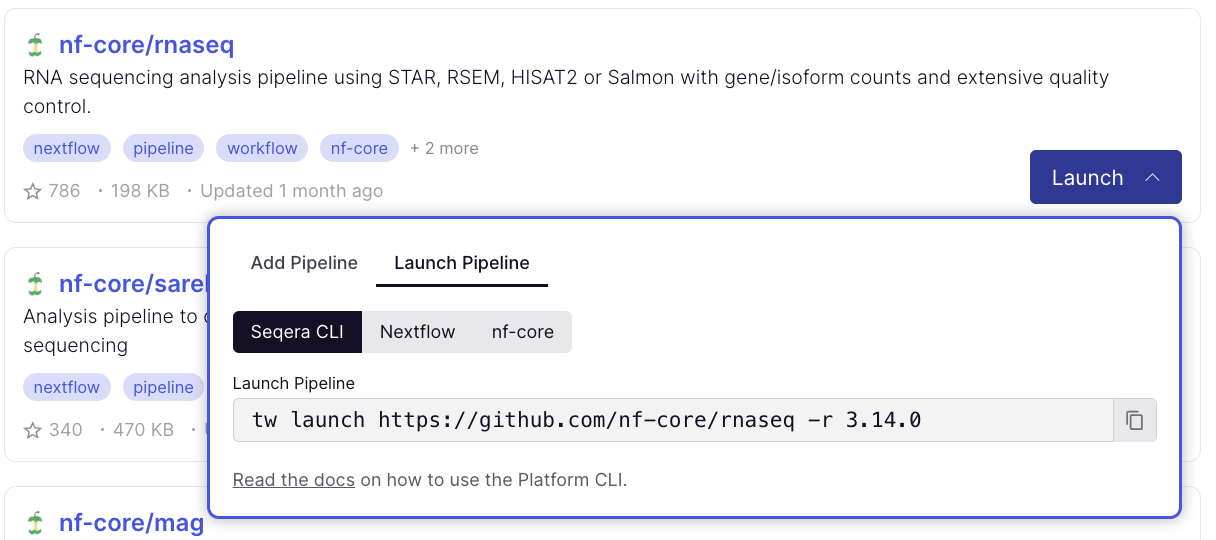
- Select Launch next to the pipeline name in the list. In the Add pipeline tab, select Cloud or Enterprise depending on your Platform account type, then provide the information needed for Seqera Pipelines to access your Platform instance:
- Seqera Cloud: Paste your Platform Access token and select Next.
- Seqera Enterprise: Specify the Seqera Platform URL (hostname) and Base API URL for your Enterprise instance, then paste your Platform Access token and select Next.
noteIf you do not have a Platform access token, select Get your access token from Seqera Platform to open the Access tokens page in a new browser window.
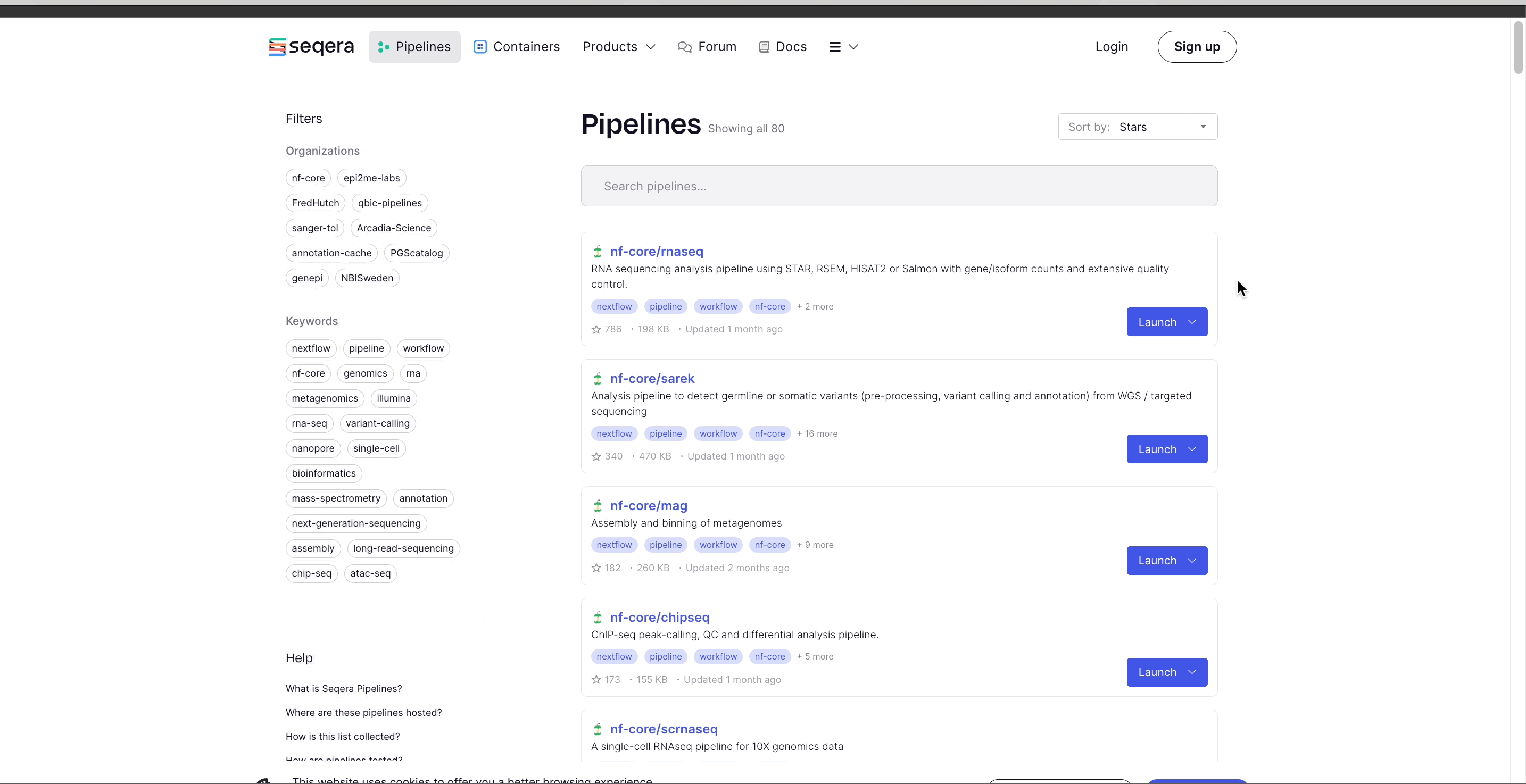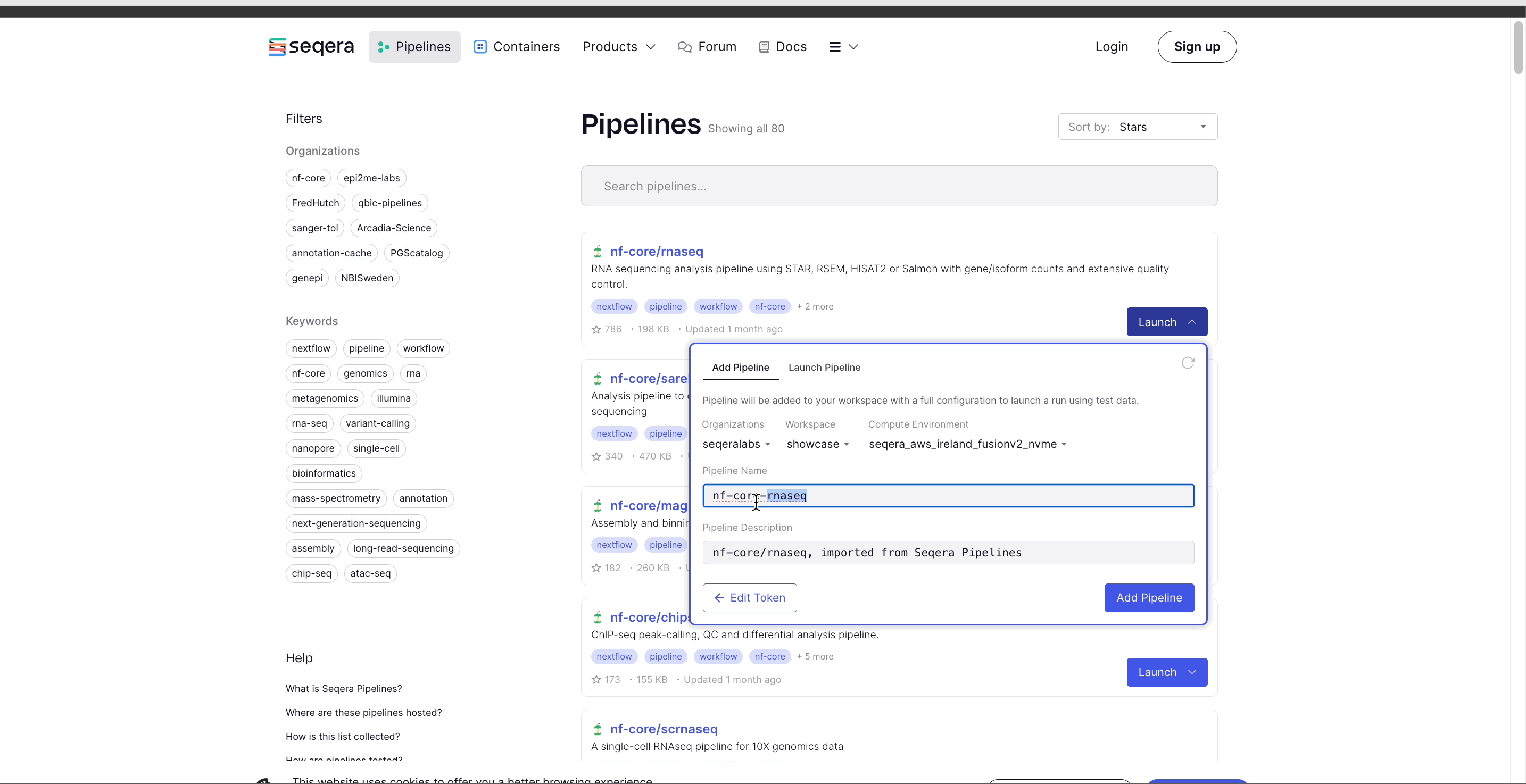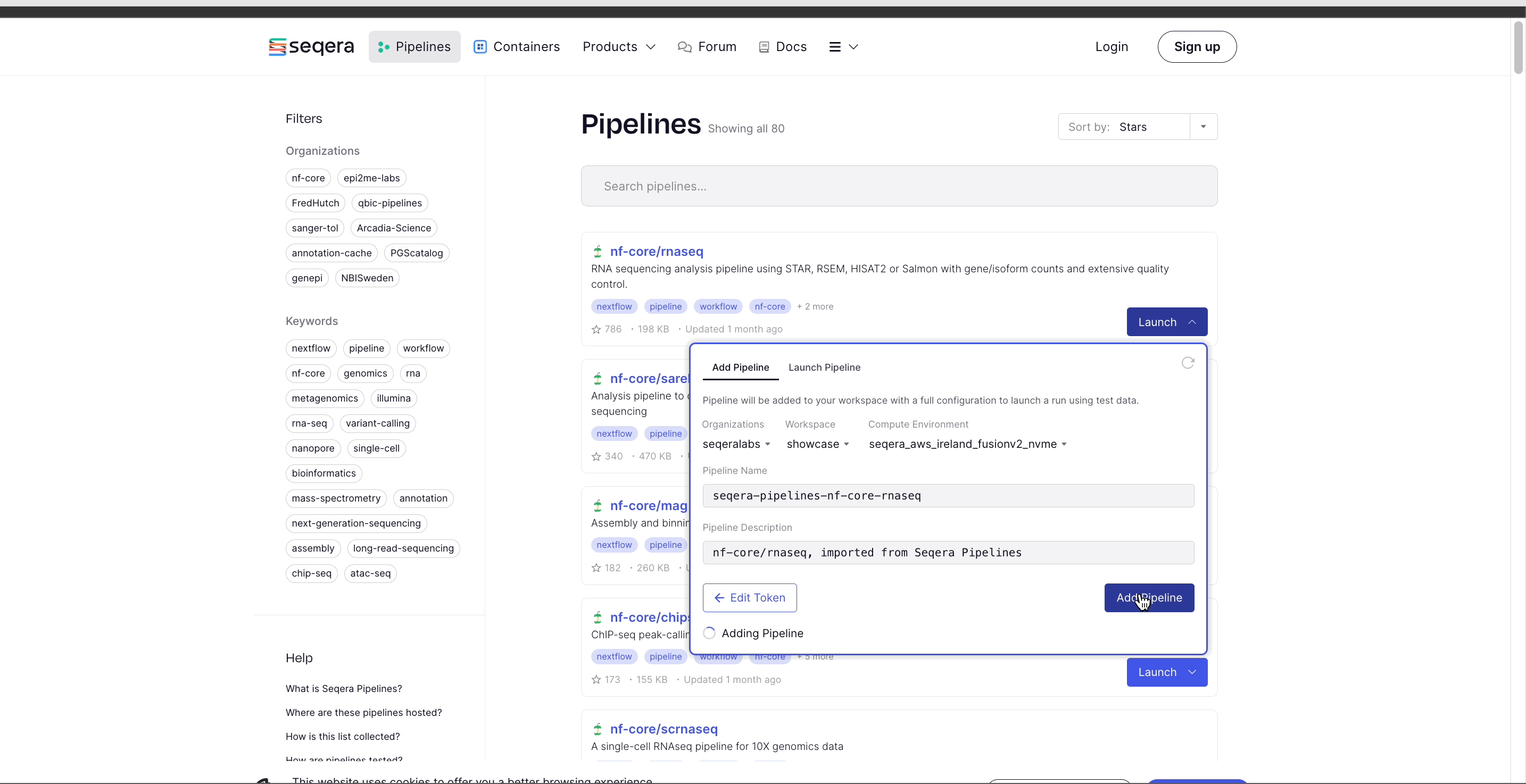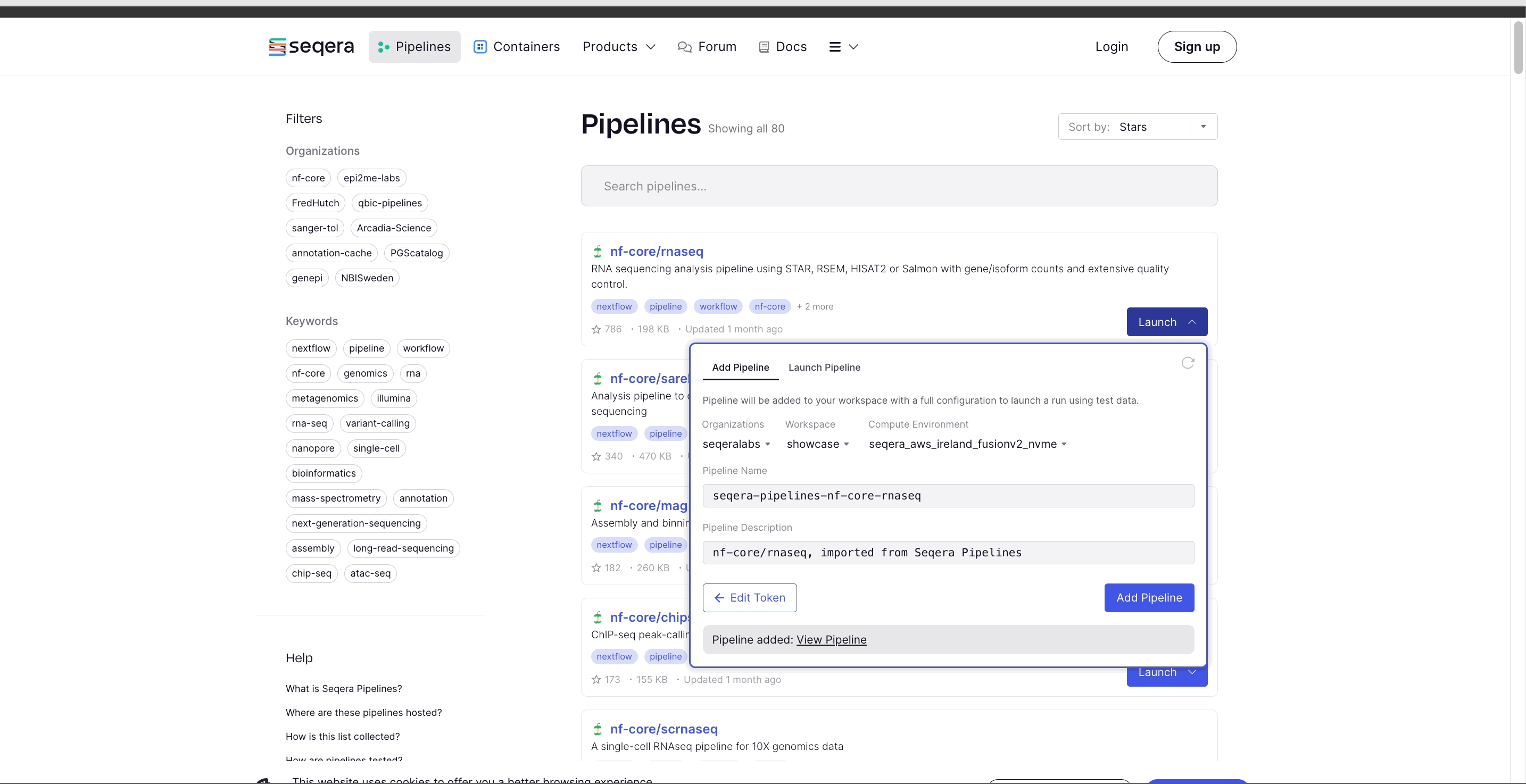
- Select the Platform Organization, Workspace, and Compute environment for the imported pipeline.
- (Optional) Customize the Pipeline Name and Pipeline Description.
note
Pipeline names must be unique per workspace.
- Select Add Pipeline.
To launch pipelines directly with CLI tools, select the Launch Pipeline tab to grab commands for Seqera Platform CLI, Nextflow, and nf-core/tools:
Add from the Launchpad
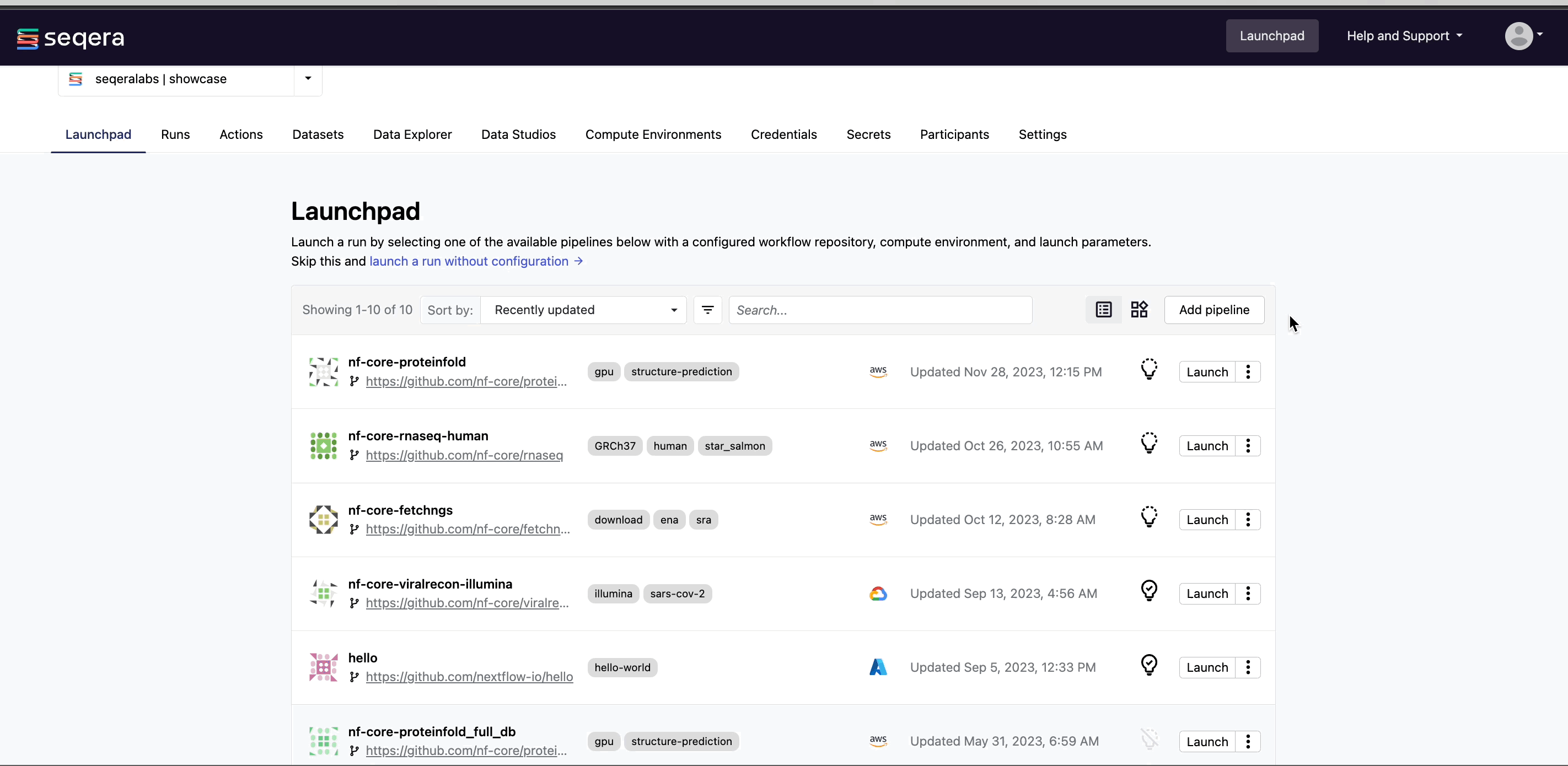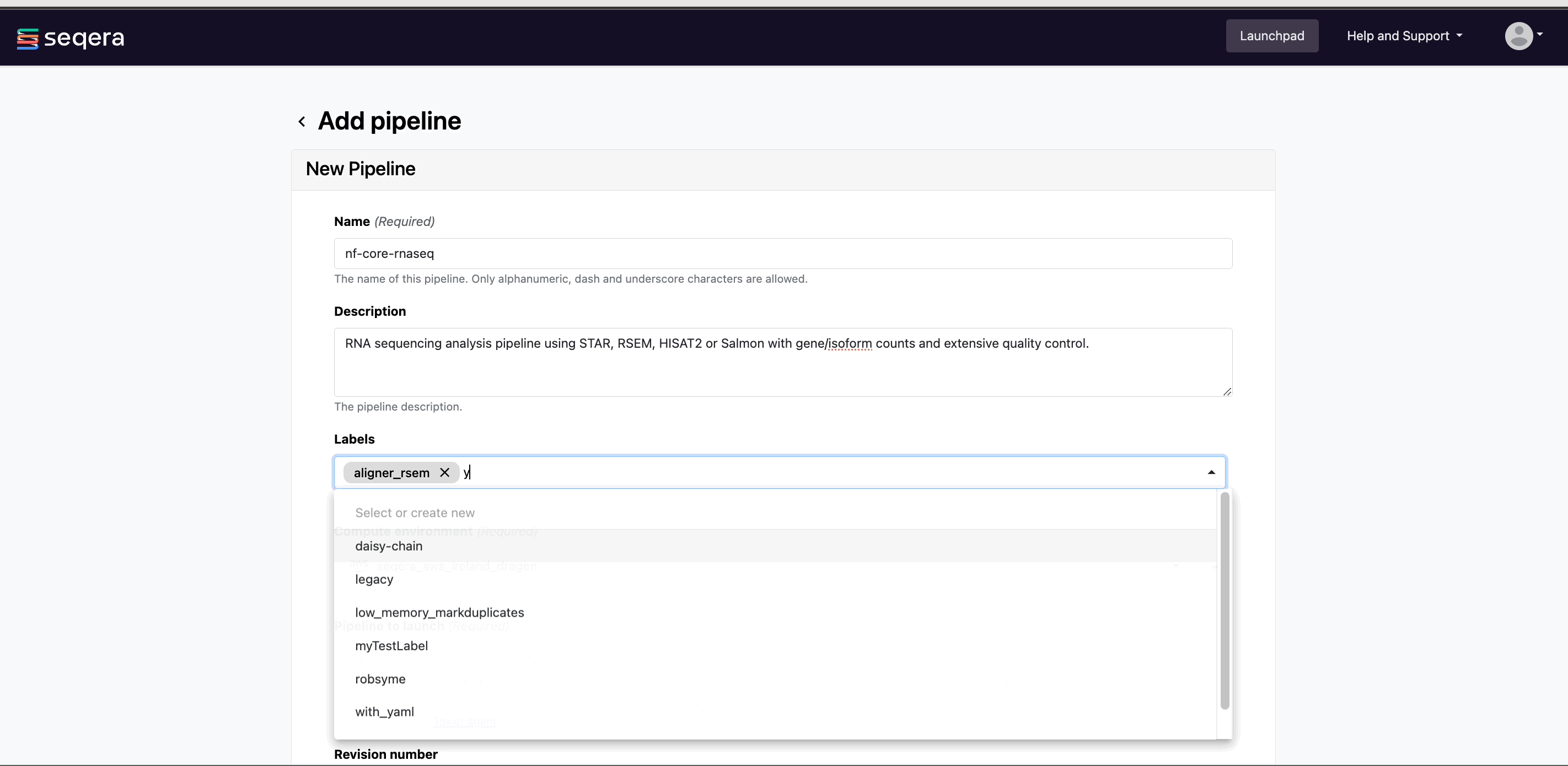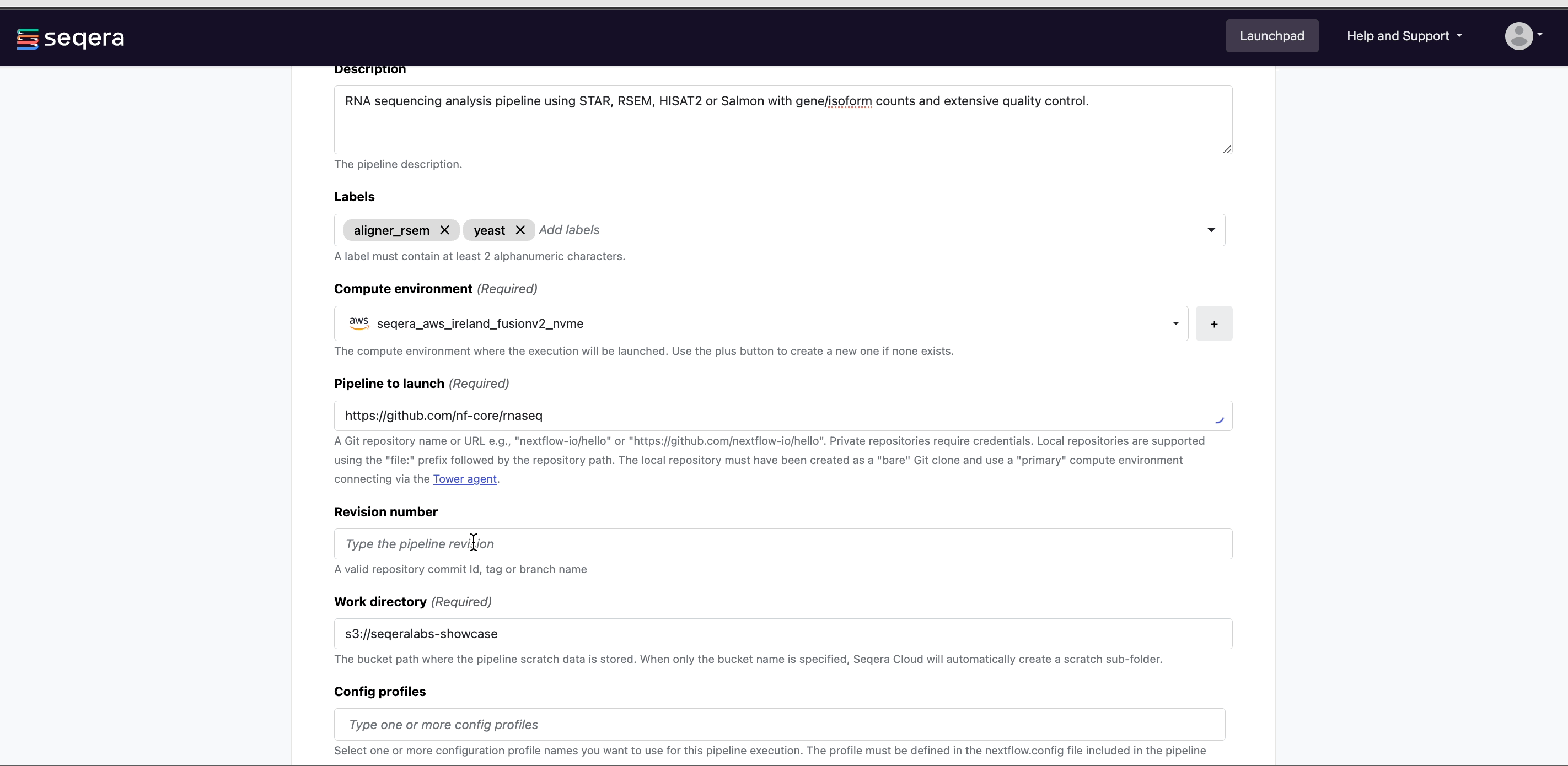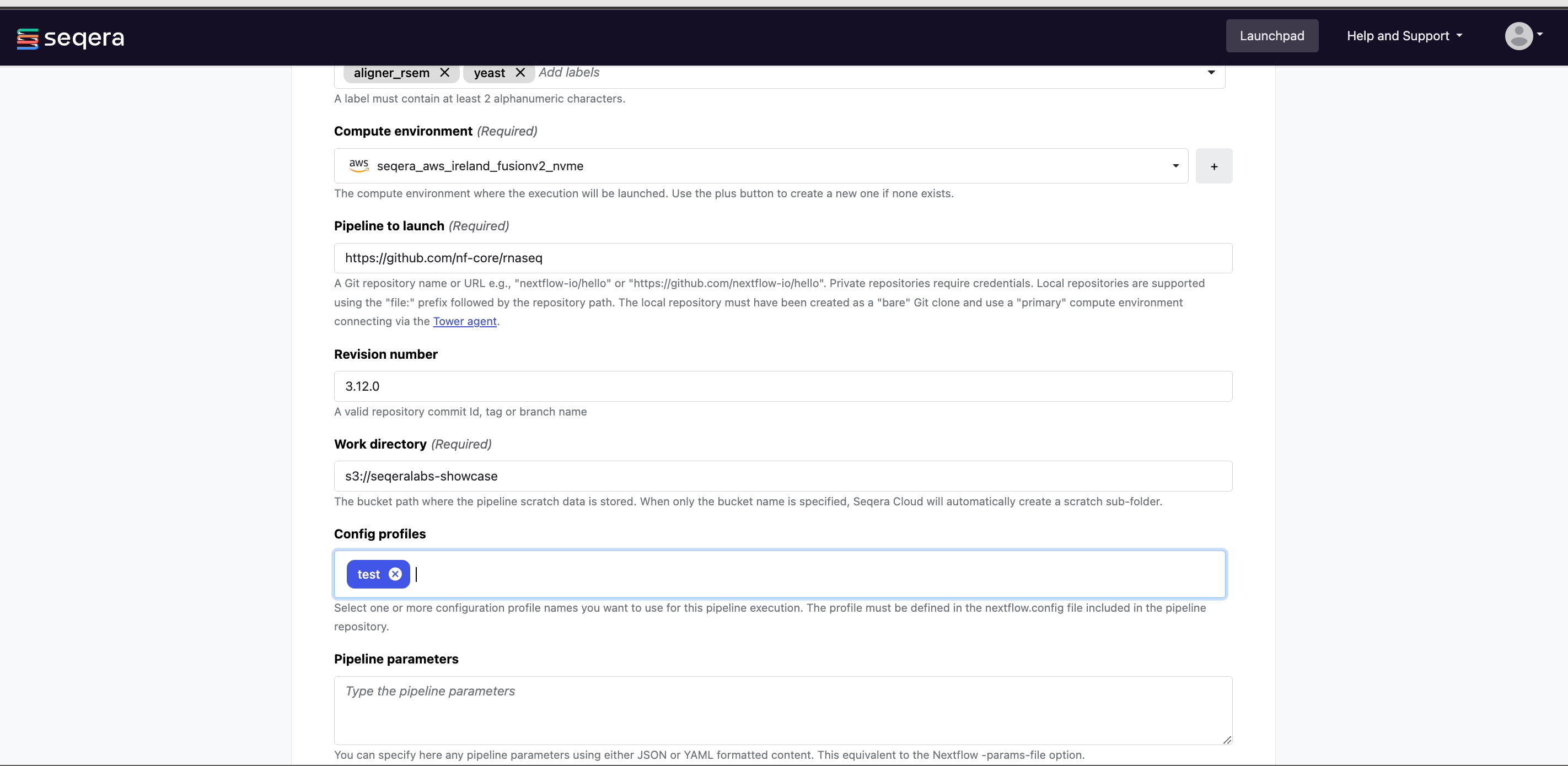
From your workspace Launchpad, select Add Pipeline and specify the following pipeline details:
-
Optional: Image: Select the Edit icon on the pipeline image to open the Edit image window. From here, select Upload file to browse for an image file, or drag and drop the image file directly. Images must be in JPG or PNG format, with a maximum file size of 200 KB.
noteYou can upload custom icons when adding or updating a pipeline. If no user-uploaded icon is defined, Platform will retrieve and attach a pipeline icon in the following order of precedence:
- A valid
iconkey:value pair defined in themanifestobject of thenextflow.configfile. - The GitHub organization avatar (if the repository is hosted on GitHub).
- If none of the above are defined, Platform auto-generates and attaches a pipeline icon.
- A valid
-
Name: A custom name of your choice. Pipeline names must be unique per workspace.
-
Optional: Description: A summary of the pipeline or any information that may be useful to workspace participants when selecting a pipeline to launch.
-
Optional: Labels: Categorize the pipeline according to arbitrary criteria (such research group or reference genome version) that may help workspace participants to select the appropriate pipeline for their analysis.
-
Compute environment: Select an existing workspace compute environment.
-
Pipeline to launch: The URL of any public or private Git repository that contains Nextflow source code.
-
Revision: A valid repository commit ID, tag, or branch name. Determines the version of the pipeline to launch.
tipSelecting a specific pipeline version is important for reproducibility as this ensures that each run with the same input data will generate the same results.
-
Commit ID: Pin pipeline revision to the most recent HEAD commit ID. If no commit ID is pinned, the latest revision of the repository branch or tag is used.
-
Pull latest: Fetch the most recent HEAD commit ID of the pipeline revision at launch time. Unpins the Commit ID, if set.
infoSee Git revision management for more information on Commit ID, Pull latest, and Revision behavior.
-
(Optional) Config profiles: Select a predefined profile for the Nextflow pipeline.
infonf-core pipelines include a
testprofile that is associated with a minimal test dataset. This profile runs the pipeline with heavily sub-sampled input data for the purposes of CI/CD and to quickly confirm that the pipeline runs on your infrastructure. -
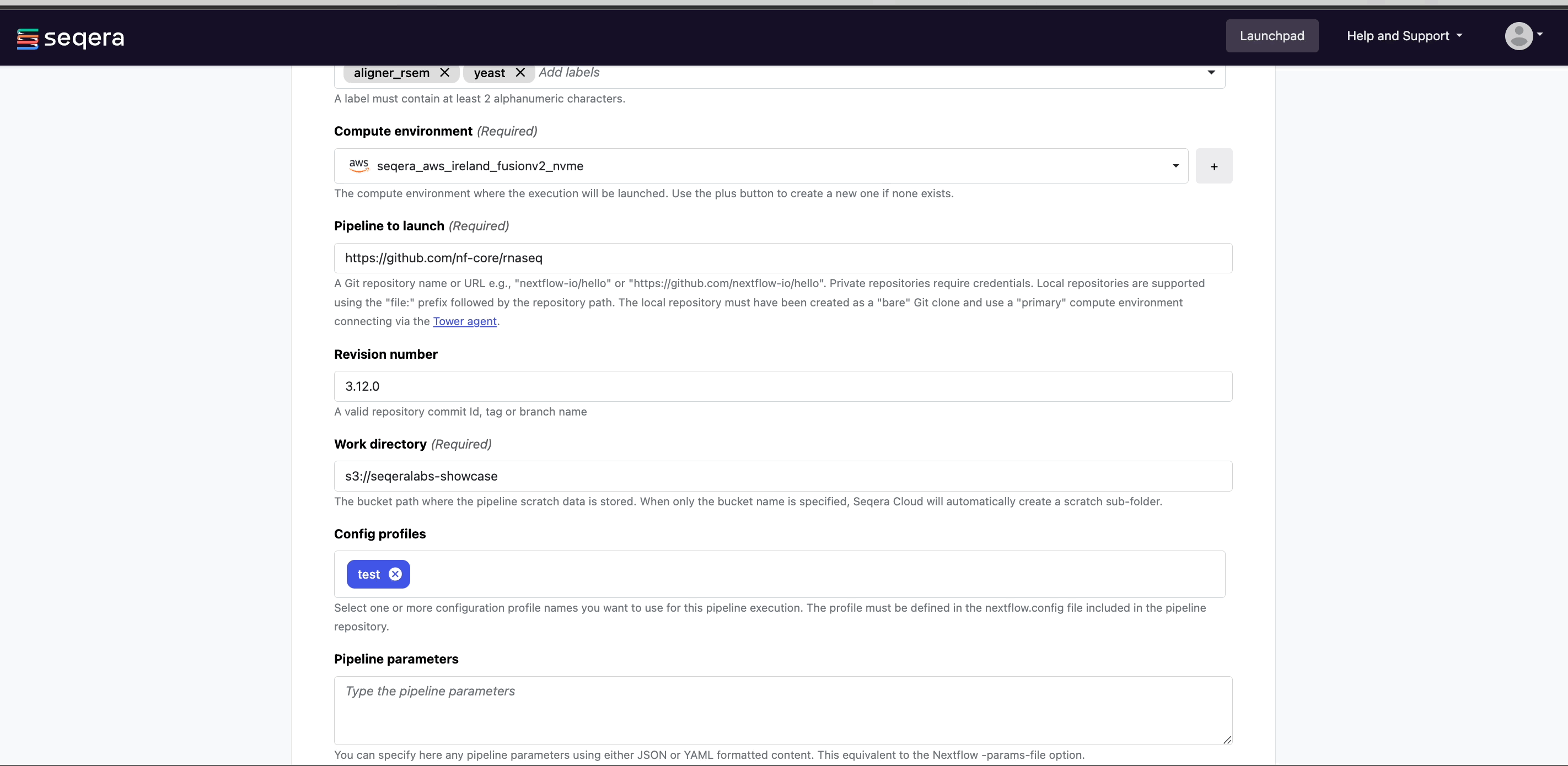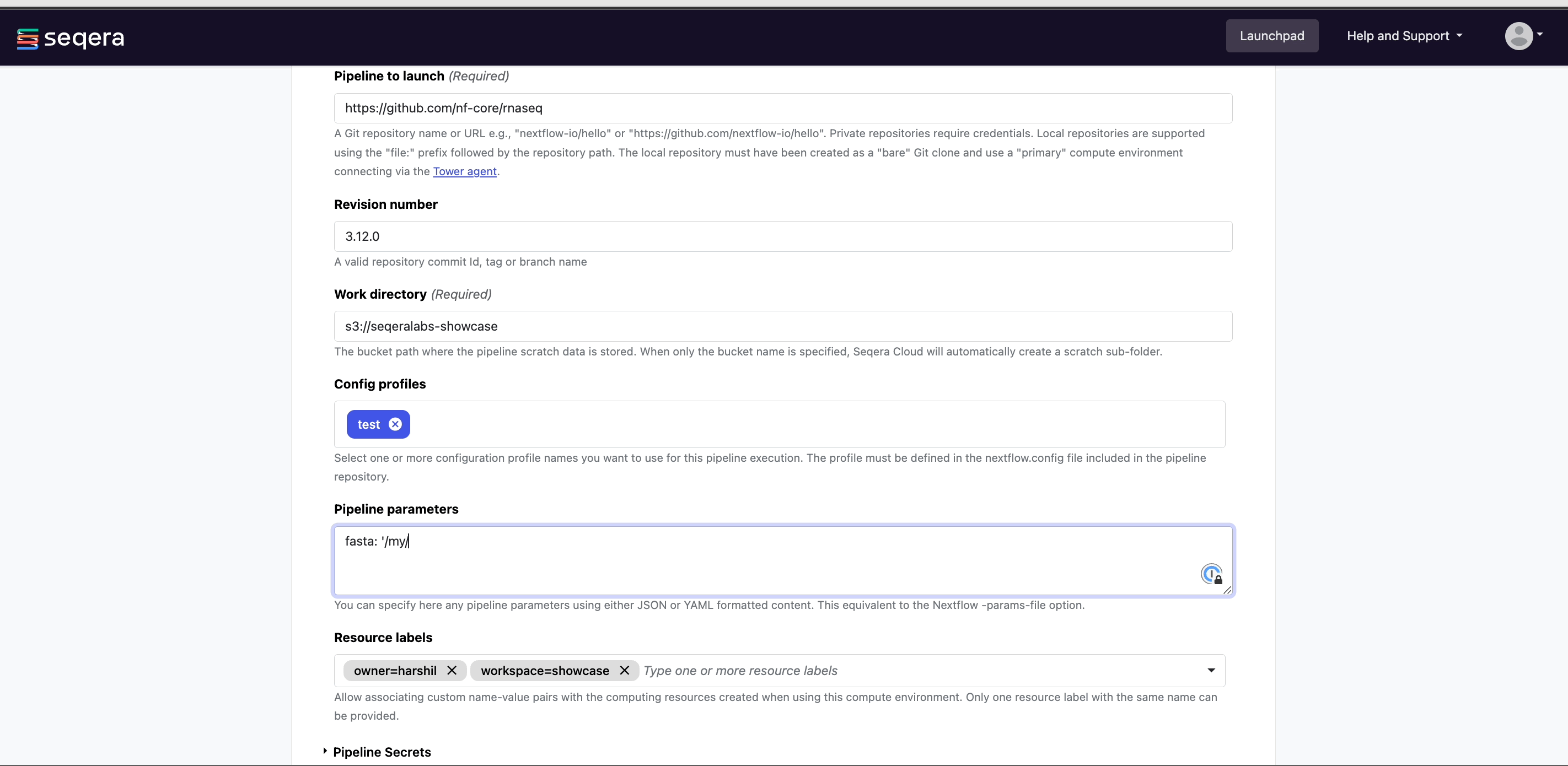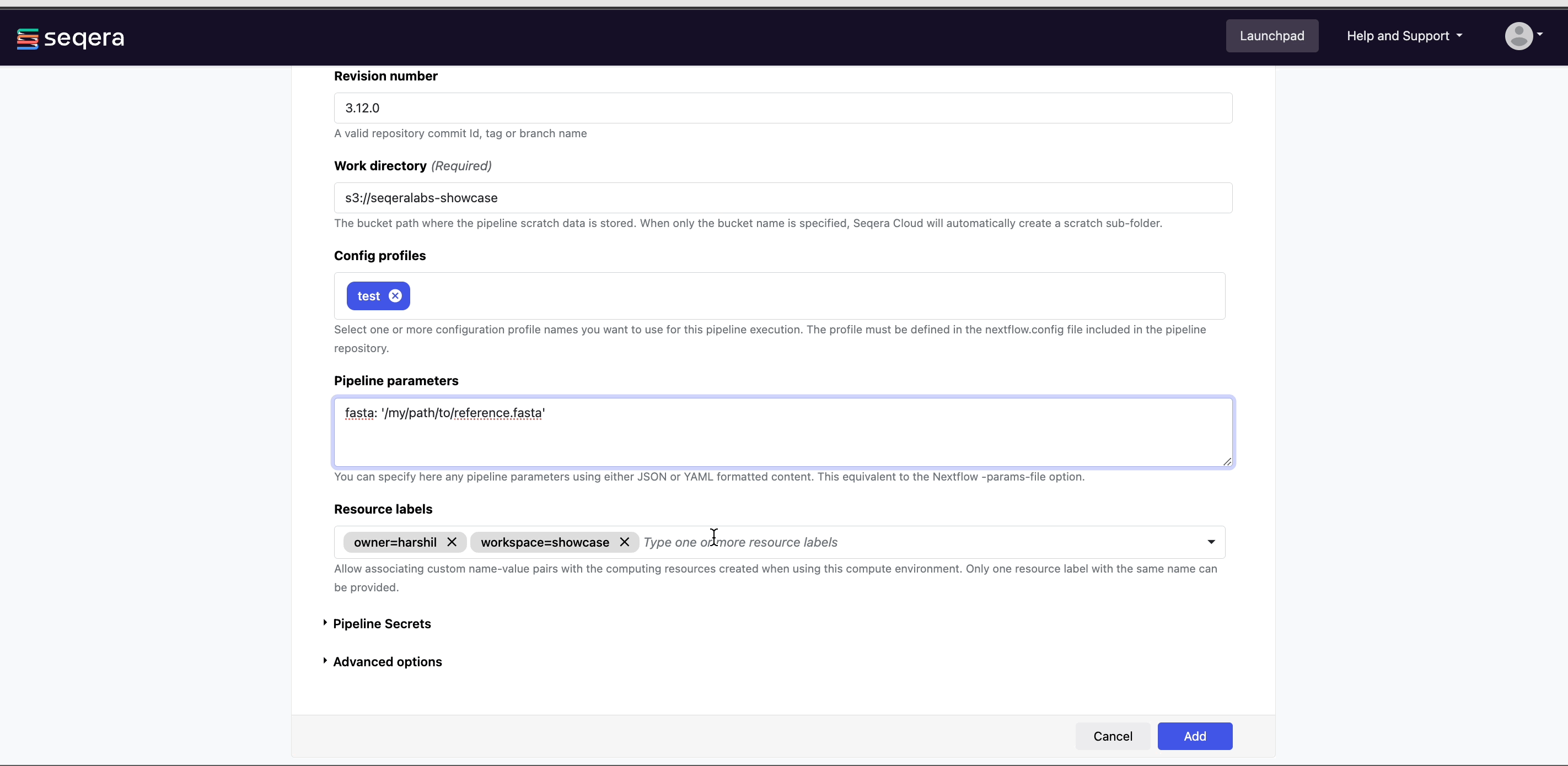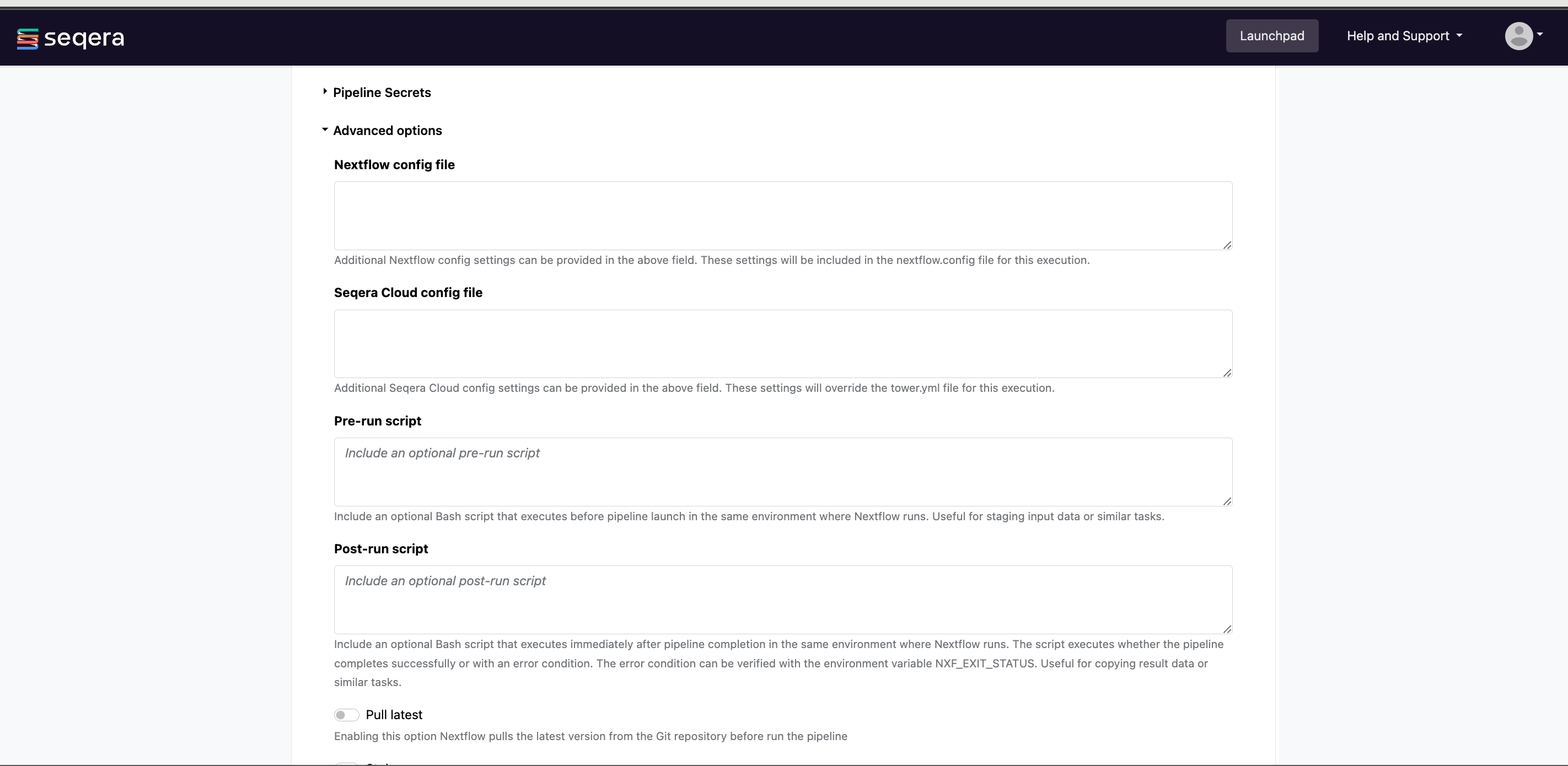
(Optional) Pipeline parameters: Set any custom pipeline parameters that will be prepopulated when users launch the pipeline from the Launchpad. For example, set the path to local reference genomes so users don't have to worry about locating these files when launching the pipeline.
-
(Optional) Pre-run script: Define Bash code that executes before the pipeline launches in the same environment where Nextflow runs.
infoPre-run scripts are useful for defining executor settings, troubleshooting, and defining a specific version of Nextflow with the
NXF_VERenvironment variable.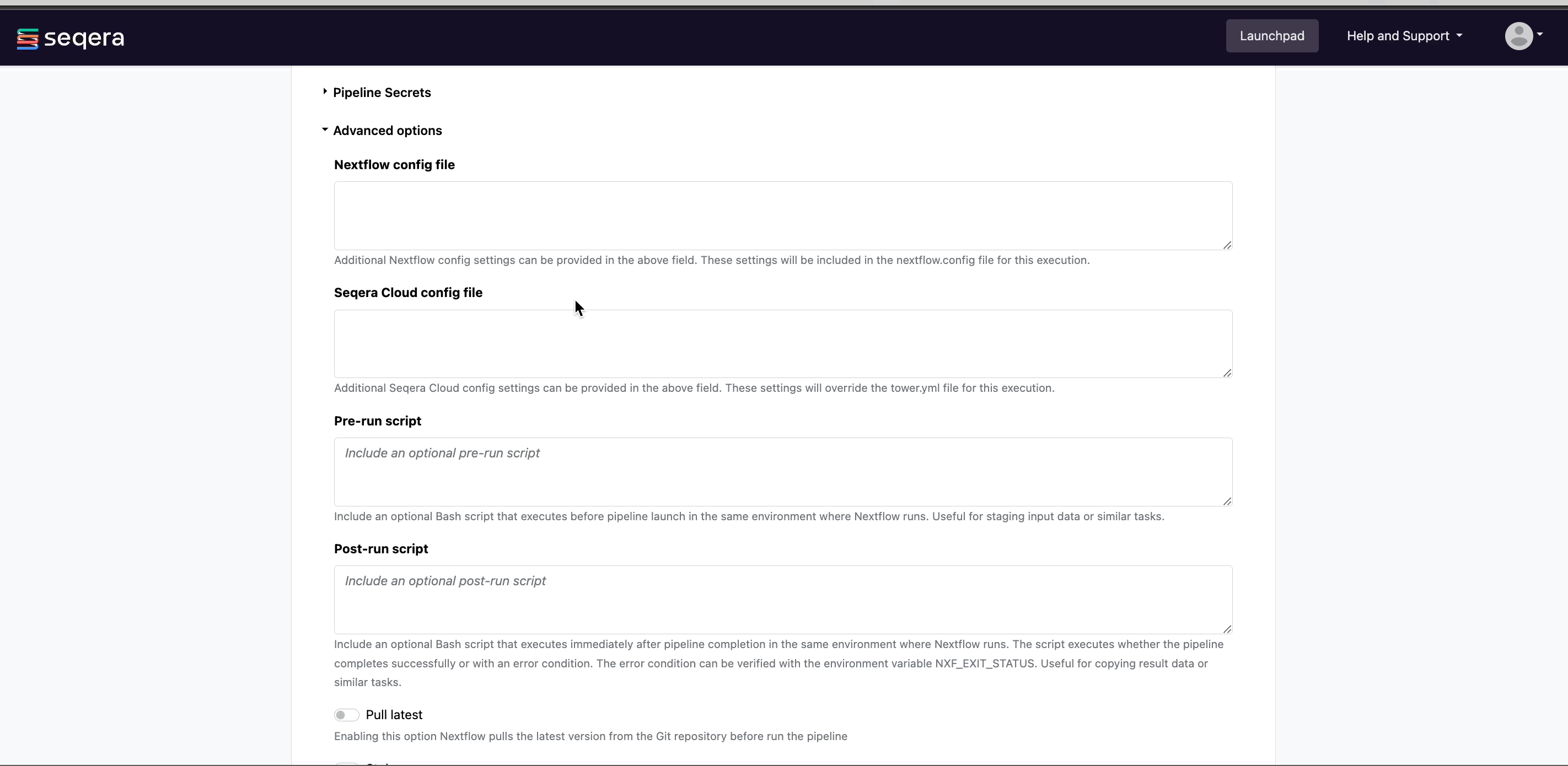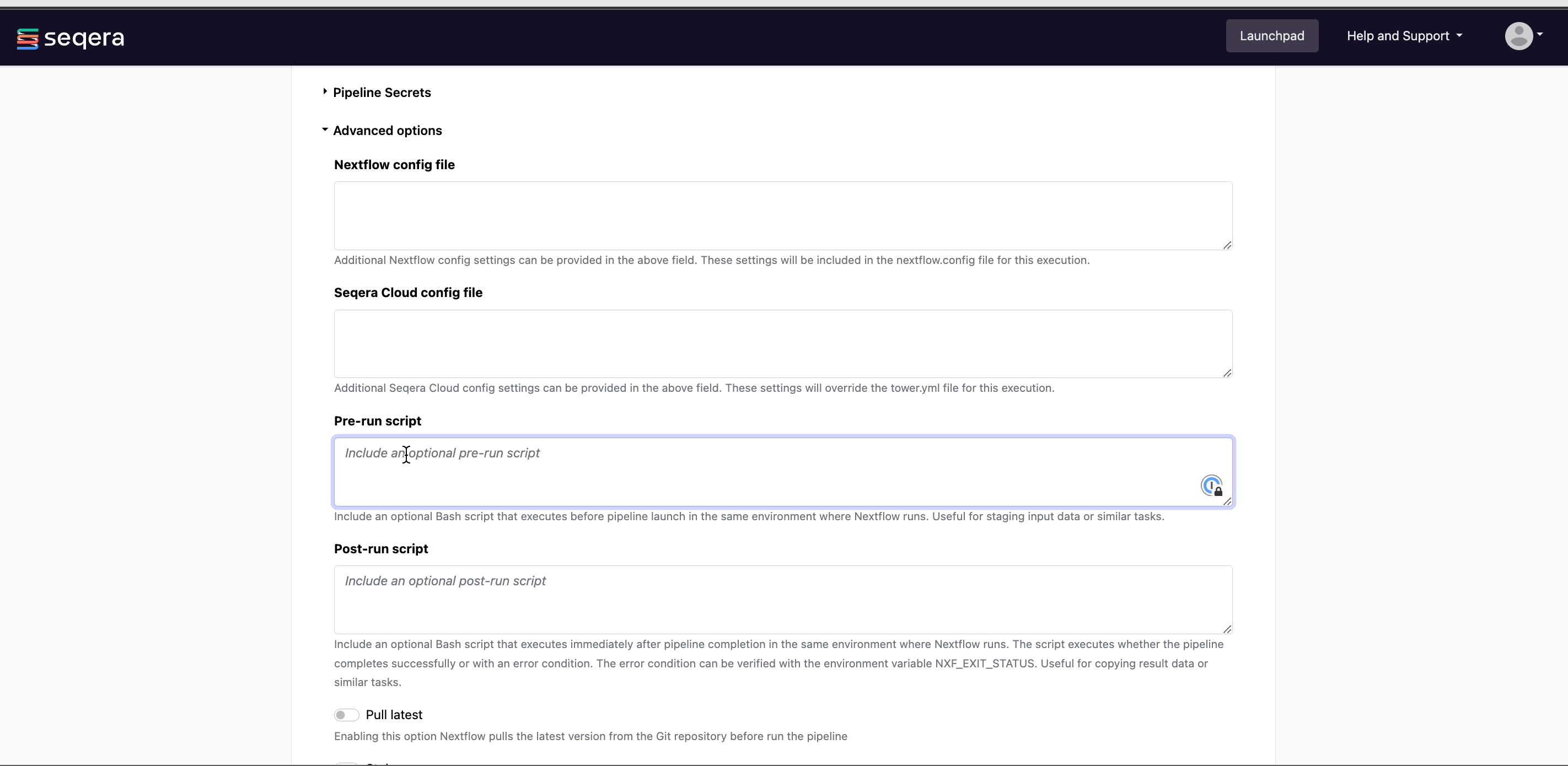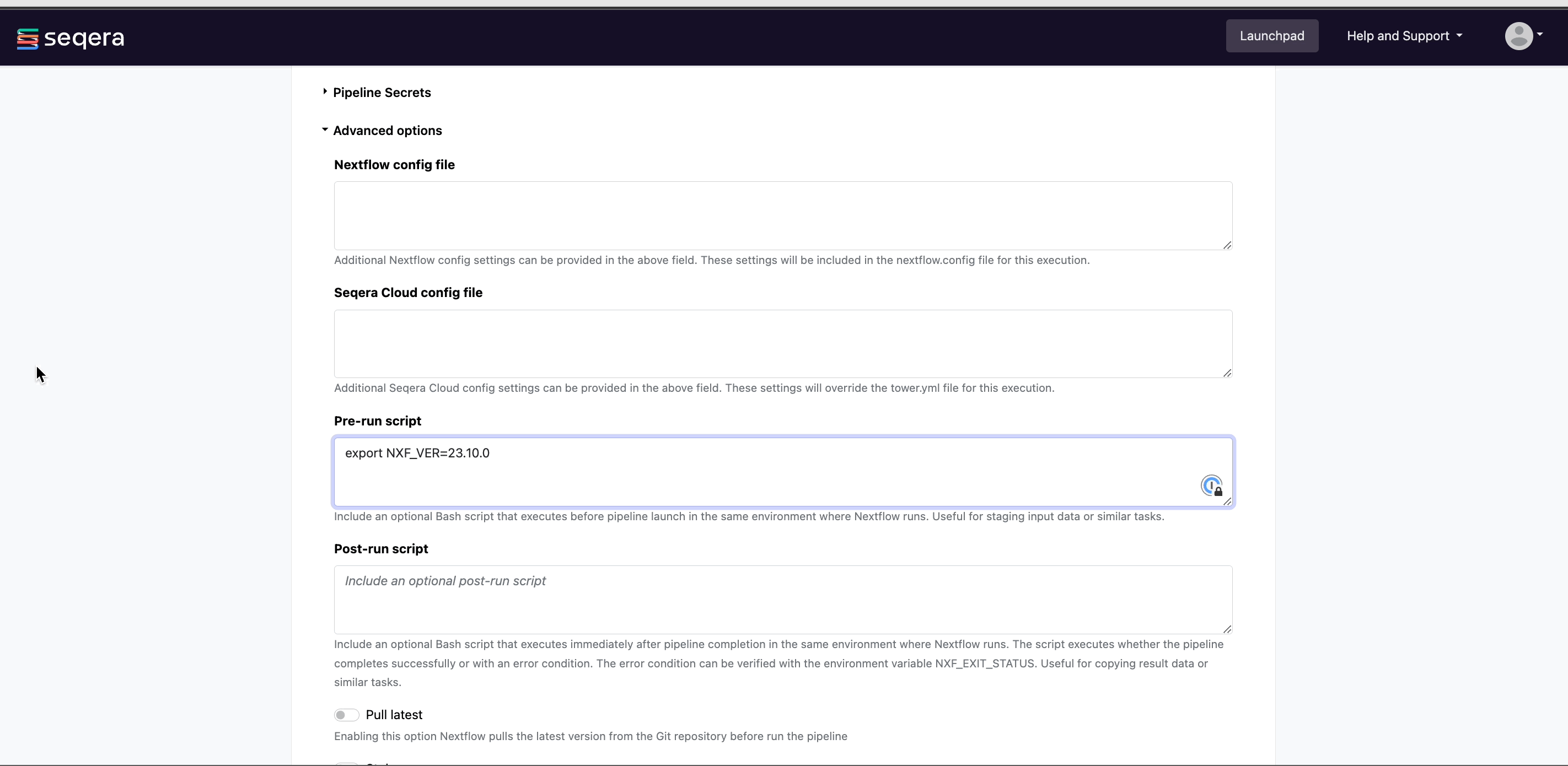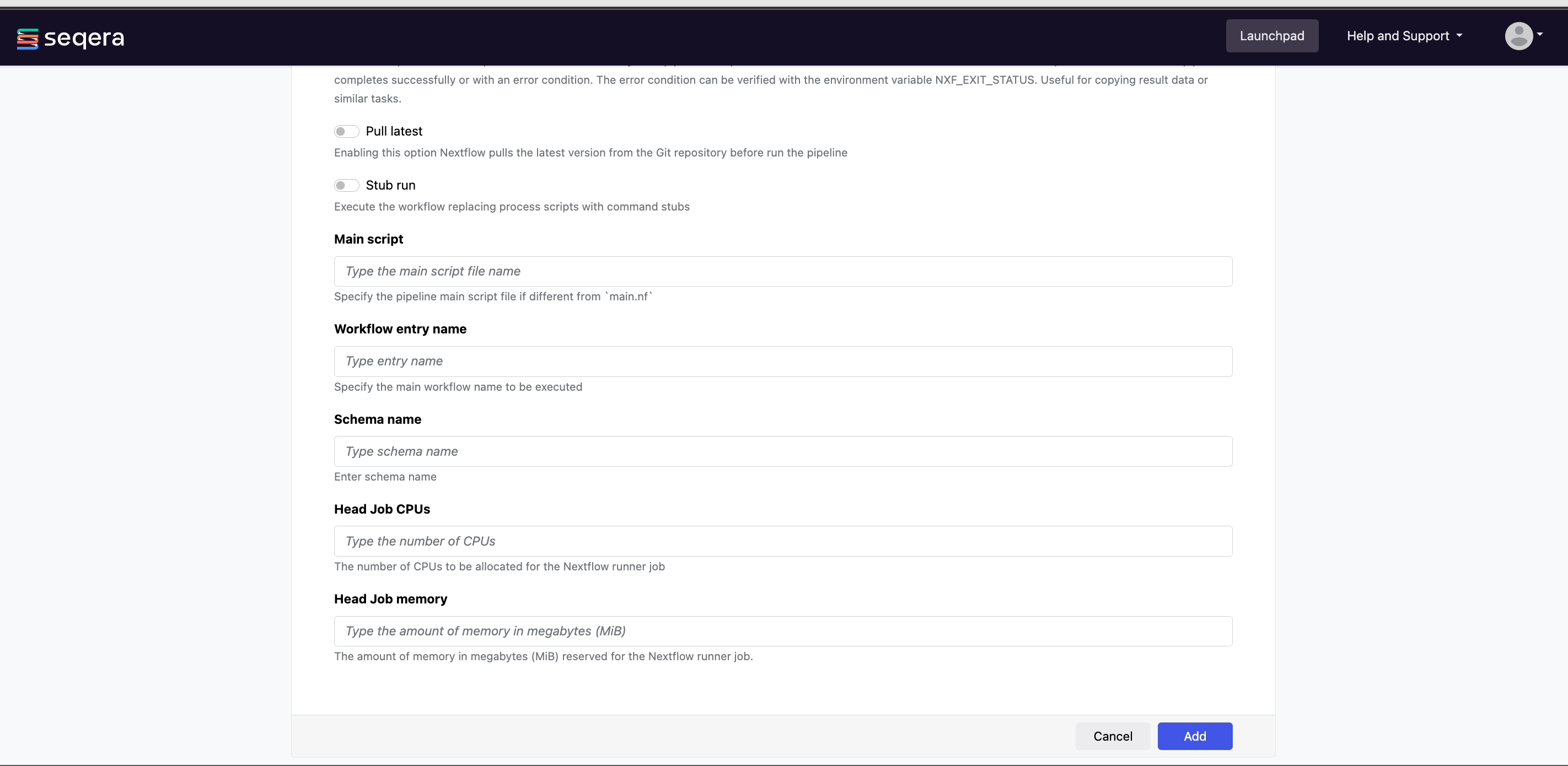
Pre-filled pipeline settings (such as compute environment, config profiles, and pipeline parameters) can be overridden during pipeline launch by workspace participants with the necessary permissions.
After you have populated the appropriate fields, select Add. Your pipeline is now available for workspace participants to launch in the preconfigured compute environment.